



















기술수출 반환과 임상실패...브릿지바이오 굴곡진 여정
- 차지현
- 2025-04-15 12:00:17
- 요약
-
가
- 가
- 가
- 가
- 가
- 가
-




- IPF 후보물질 BBT-877, 2상 탑라인 결과 1차 평가지표 미충족
- 2017년 리가켐서 도입, 2019년 베링거에 1조원대 L/O 후 반환
- "전체 데이터 수령 후 추가 분석…부작용 문제 없어, 추가 적응증 검토"
- PR
- 8/7 물갈이, 배탈 등 여름철 장질환 온라인세미나
- 사전 신청하기

15일 금융감독원에 따르면 브릿지바이오테라퓨틱스는 14일 특발성 폐섬유증(IPF) 치료제 후보물질 'BBT-877' 다국가 임상 2상 탑라인(주요 지표) 데이터 분석 결과 1차 평가지표인 24주차 강제 폐활량(FVC) 변화에서 유의미한 개선 효과를 확인하지 못했다.
BBT-877 임상 2상은 IPF 환자를 대상으로 약물의 유효성과 안전성, 내약성을 평가하기 위해 한국, 미국, 호주, 폴란드, 이스라엘 등 5개국에서 진행됐다. 총 129명의 환자가 참여했다. 연구 결과 FVC 변화가 약물군과 위약군 모두에서 관찰됐으나, 두 군 간 통계적 유의미한 차이는 없었다.
IPF는 알 수 없는 이유로 섬유 조직이 과도하게 생성되면서 폐가 딱딱하게 굳어가는 질환이다. 진단 후 5년 생존율이 40% 미만인 희소질환이다. 2023년 기준 전 세계 IPF 환자 수는 약 30만명에서 50만명으로 추정된다. 이 중 미국에만 약 10만명 이상 환자가 있는 것으로 알려진다.
현재 미국 식품의약국(FDA) 승인을 받은 IPF 치료제는 베링거인겔하임의 '오페브'(성분명 닌테다닙)와 로슈의 '에스브리엣'(성분명 퍼페니돈) 2개 품목밖에 없다 각각 2023년 매출이 7억7000만달러와 37억달러에 달한다. 다만 두 약물 모두 폐기능 저하를 늦출 뿐, 근본적인 치료 가능성이 낮아 미충족 의료 수요가 높은 상황이다.
앞서 브릿지바이오는 2017년 5월 리가켐바이오로부터 BBT-877에 대한 전 세계 독점실시권을 획득했다. 브릿지바이오는 반환 의무가 없는 선급금(업프론트) 20억원을 포함해 총 300억원에 BBT-877의 권리를 도입했다. 계약에는 브릿지바이오가 기술수출을 달성할 경우 이후 개발·판매 과정에서 단계별로 발생하는 모든 수익 중 45%를 리가켐바이오와 배분하는 조건이 달렸다.
브릿지바이오는 2019년 7월 독일 베링거인겔하임에 BBT-877을 최대 1조5000억원 규모로 기술수출했다. 임상 1상 단계의 BBT-877을 넘기면서 업프론트와 단계별 기술료(단기 마일스톤)으로 약 600억원을 받았다. 2019년 말 BBT-877 임상 1상 완료에 따라 약 50억원을 마일스톤 수익분배금 명목으로 리가켐바이오에 지급했다.
그러나 이듬해인 2020년 베링거인겔하임은 잠재적 독성 문제로 BBT-877의 권리를 반환했다. 브릿지바이오는 추가 실험에서 독성 문제가 고농도 약물 처리에 따른 것으로 판단, 자체적으로 후보물질을 개발하기로 결정했다. BBT-877 원개발사 리가켐바이오는 2021년 초 제3자배정 방식 유상증자를 통해 브릿지바이오에 50억원 규모 자금을 지원하면서 개발 지속에 힘을 보탰다.
브릿지바이오는 2022년 5월 BBT-877의 안전성을 다각도로 입증할 수 있는 추가 자료를 FDA에 모두 제출했다고 밝혔다. 회사가 제출한 자료에는 약물 복용 후 DNA 손상 원인이 약물 자체의 유전 독성에 의한 직접적인 손상이 아닌, 고농도 약물 처리에 의한 간접적인 손상에 있다는 혜성분석 실험 결과가 포함됐다.
이후 브릿지바이오는 2022년 7월 FDA로부터 BBT-877 임상 2상 임상시험계획(IND)을 승인받았다. 이어 2023년 4월 호주에서 첫 환자 투약을 시작했다. 브릿지바이오는 대한민국, 폴란드, 이스라엘 등에서 임상을 진행해 왔다.
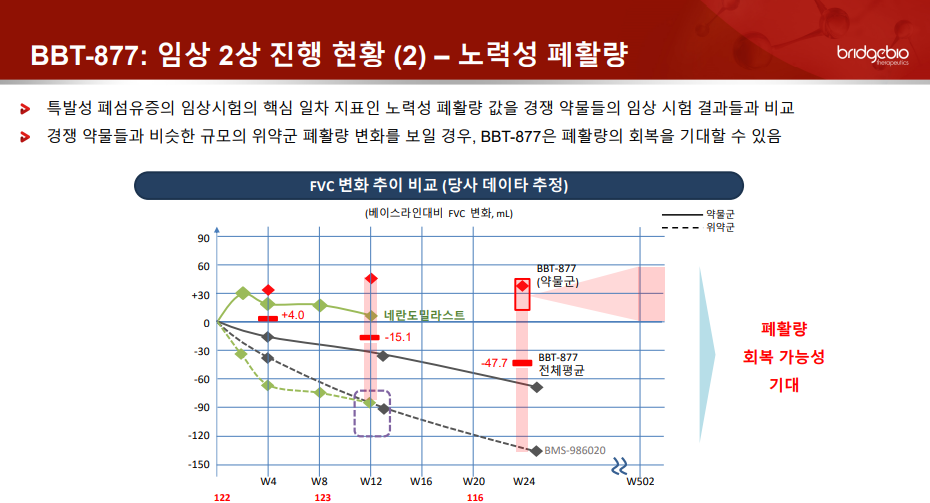
브릿지바이오는 올 초 미국 샌프란시스코에서 열린 JP모건 헬스케어 콘퍼런스(JPM)에서 국내 신약개발 바이오텍 가운데 처음으로 메인 세션 발표 업체로 선정돼 BBT-877 최신 임상 2상 진행 현황을 공개한 바 있다. 또 브릿지바이오는 JPM에서 1000명 이상 IPF 환자를 대상으로 52주간 투약하는 BBT-877 임상 3상 연구 계획과 기술수출 계획 등을 구체적으로 공개했다.
당시 브릿지바이오 측은 "지난 12월 말 기준 BBT-877 치료군과 위약군 105명 피험자에서 확인된 FVC 전체 평균 변화값은 투약 24주 후 측정 기준 -44.3mL로, 기존 경쟁 약물 임상들에서의 위약군 피험자들이 보인 폐활량 감소폭(약 -104 ~ -134mL)을 고려했을 때 BBT-877 투약군 피험자의 폐기능 회복 가능성이 기대된다"고 했다.
또 회사 측은 "이번 JPM 행사에서 다수 글로벌 빅파마와 BBT-877 기술수출 계약 타진을 위한 구체적 협의도 진행했다"면서 "현재까지 진행된 사업개발 활동을 통해 글로벌 상위 10개 빅파마 중 절반이 넘는 다수의 기업과 기밀유지협약(CDA)을 체결했고 JPM 현장에서 BBT-877 향후 임상 계획 등을 기반으로 기술거래 방식 및 세부 일정 등 구체적인 사항을 논의했다"고도 덧붙였다.

이 대표는 "MMRM 통계 모델에는 기저 FVC 수치, 나이, 성별 등 인체 특성, 측정값이 빠진 환자까지 고려해서 수치를 보정한다"면서 "이런 보정 작업을 하면, 실제보다 변화량이 더 크게 나타날 수도 있고 이 같은 통계처리 모델 때문에 나오는 수치의 차이가 있다는 걸 이해해달라"고 했다.
이번 임상 결과로 브릿지바이오의 BBT-877 임상 3상과 기술수출 계획에 차질이 생기게 됐다. 회사는 최종 임상시험 결과보고서를 수령한 후 하위 그룹 분석, 바이오마커 결과와 고해상도 CT 영상 분석 등을 통해 이번 결과를 포함한 개별 환자 데이터를 면밀히 검토할 예정이다. 이를 바탕으로 향후 임상 개발과 사업 전략을 재수립하겠다는 계획도 내놨다.
향후 계획과 관련 이 대표는 "현재는 BBT-877 탑라인만 받은 상황이라 전체 데이터를 받은 뒤 추가 분석을 통해 세부사항을 확인하는 걸 우선으로 할 계획"이라며 "당초 우려했던 부작용 프로파일은 별다른 문제가 없는 것으로 발견됐으므로 BBT-877 추가 적응증을 적극 검토할 예정"이라고 했다.
이 대표는 "원개발자인 리가켐바이오와 면밀히 협의해 향후 대응 방안을 같이 모색할 것"이라면서 "현재 탑라인 데이터로는 BBT-877의 즉각적인 기술이전이 쉽지 않은 게 사실이지만 추가 분석 데이터 등을 기반으로 빅파마와 논의를 재개하고 기술수출 가능성을 알아보겠다"고도 덧붙였다.
- 익명 댓글
- 실명 댓글
- 댓글 0
- 최신순
- 찬성순
- 반대순
-
 김천에서 정규직약사님모셔요 5:30퇴근,급여740(퇴직금선지급시실수령800)
김천에서 정규직약사님모셔요 5:30퇴근,급여740(퇴직금선지급시실수령800) -
 한국에자이 Market Access Specialist 채용
한국에자이 Market Access Specialist 채용 -
 약제팀 (협약직) 야간약사 모집
약제팀 (협약직) 야간약사 모집 -
 NPP & BD (Sr.) Specialist, CSE&BD (Fixed)
NPP & BD (Sr.) Specialist, CSE&BD (Fixed) -
(Sr.) MSL, Hematology (Permanent)
-
 상주적십자병원 계약직 약사 모집 공고
상주적십자병원 계약직 약사 모집 공고 -
 충청지역 의원 영업 팀장 채용
충청지역 의원 영업 팀장 채용 -
 근무 약사 채용(풀 타임)
근무 약사 채용(풀 타임) -
 은평성모병원 2026년도 하반기 약사 공개채용(시간제, 야간전담)
은평성모병원 2026년도 하반기 약사 공개채용(시간제, 야간전담) -
 [강동성심병원] 주말약사 모집공고
[강동성심병원] 주말약사 모집공고 -
 [한독] 신입 및 경력 직무별 수시채용
[한독] 신입 및 경력 직무별 수시채용 -
 [일산백병원] 약제부 계약직 야간/주말당직약사 (신입/경력) 모집 공고
[일산백병원] 약제부 계약직 야간/주말당직약사 (신입/경력) 모집 공고 -
 [제뉴원사이언스] 품질관리약사 모집(경력무관)
[제뉴원사이언스] 품질관리약사 모집(경력무관) -
Senior Manager, Government Affairs & External Liaison (Permanent)
-
 토요일 당직약사님 모집
토요일 당직약사님 모집 -
 [윌스기념병원] 약제팀 약사 초빙
[윌스기념병원] 약제팀 약사 초빙 -
 국립과학수사연구원 약사 3명 채용
국립과학수사연구원 약사 3명 채용 -
 임상PM/제제개선/건기식개발담당 채용
임상PM/제제개선/건기식개발담당 채용 -
 향남공장 OQA 품질약사 채용(주5일/파트타임 가능)
향남공장 OQA 품질약사 채용(주5일/파트타임 가능) -
 의정부/노원을지대학교병원 약사 채용
의정부/노원을지대학교병원 약사 채용 -
 세종공장 품질관리약사(사원~과장)
세종공장 품질관리약사(사원~과장) -
 동성제약 아산공장 약사 채용
동성제약 아산공장 약사 채용 -
 병원영업(MR) 채용연계형 인턴(신입사원) 모집 공고
병원영업(MR) 채용연계형 인턴(신입사원) 모집 공고 -
 [서초구 종합병원] 기쁨병원에서 약사 초빙합니다.
[서초구 종합병원] 기쁨병원에서 약사 초빙합니다. -
약제팀 (정규직) 약사 모집
-
 [SK바이오팜] 각 부문 신입/경력 구성원 영입
[SK바이오팜] 각 부문 신입/경력 구성원 영입 -
 [H+양지병원] 약제팀 주간 약사 모집 (정규직)
[H+양지병원] 약제팀 주간 약사 모집 (정규직) -
 JW 2026년 4차 수시채용
JW 2026년 4차 수시채용 -
 본사 사업개발 팀원&팀장 채용
본사 사업개발 팀원&팀장 채용 -
 순천향대학교 부속 구미병원 약제팀 계약직 야간약사 채용공고
순천향대학교 부속 구미병원 약제팀 계약직 야간약사 채용공고 -
 2026년 국립소방병원 약무직 신규직원 채용 공고
2026년 국립소방병원 약무직 신규직원 채용 공고 -
 평택공장 제조관리약사 채용(신입우대)
평택공장 제조관리약사 채용(신입우대) -
 [대자인병원] 약제팀 약사 / ASP팀 감염전문약사 모집
[대자인병원] 약제팀 약사 / ASP팀 감염전문약사 모집

약국e몰
![[신신제약] 모스키토 밀크](https://cdn.platpharm.co.kr/2025/10/2510150733400004067.webp)
![[유한양행] 안티푸라민 파스 시리즈](https://cdn.platpharm.co.kr/2024/05/2405280631070000069.png)
![[옵투스] 오에수 시리즈](https://cdn.platpharm.co.kr/2026/02/2602130209000031633.webp)
![[일양약품] 프로엑스피](https://cdn.platpharm.co.kr/2026/01/2601221008450010125.webp)
![[삼진제약] 게보핏 시리즈](https://cdn.platpharm.co.kr/2024/07/2407100728250000386.png)
![[동성제약] 정로환 F정](https://cdn.platpharm.co.kr/static/dailypharm/jeongrohwan-f.png)
![[일양약품] 도담도담 시리즈](https://cdn.platpharm.co.kr/2024/02/2402020935180000240.jpg)
![[종근당] 브레이닝캡슐](https://cdn.platpharm.co.kr/2025/06/2506040708450012544.png)
![[리쥬올] PDLLA 퍼밍 크림 30ml](https://cdn.platpharm.co.kr/2026/04/2604070229110000386.webp)
![[노보노디스크] 위고비](https://cdn.platpharm.co.kr/static/dailypharm/Wigobi.png)
![[신신제약] 아렉스마일드](https://cdn.platpharm.co.kr/2023/11/2311300927130000133.jpg)
![[경방신약] 방콜브이산](https://cdn.platpharm.co.kr/2025/12/2512310630020002495.webp)
![[켄뷰] 다양한 통증에, 타이레놀정 500mg 10정](https://i.baropharm.com/products/6c6ea4f4-7ab2-44f2-a165-f062d80f525b.png)
![[휴온스 ] 비듬을 한번에, 니조랄 2%액](https://i.baropharm.com/products/478a284d-4361-4b4a-8a00-8bab80f34319.png?label=PLAN_01)
![[쥬베룩] 진짜 쥬베룩을 담은 약국전용 PDLLA 크림](https://i.baropharm.com/products/202604/1775343960671.png?label=바뷰페로고)
![[여드름치료]아크스팟크림](https://i.baropharm.com/products/202607/1782883852617.png)
![[리쥬올] 닥터 리쥬올 어드밴스드 PDRN 리쥬비네이팅 크림 30ml](https://i.baropharm.com/partner/products/a201d2b4-f21e-4b13-957c-846d286b3d21.jpg?label=바뷰페로고)
![[아워팜] 우리아이 맞춤설계, 바로타민 kids 엘더베리맛](https://i.baropharm.com/partner/products/3f39593e-6318-4dd9-a778-c008c868b5c8.png)
![[켄뷰] 오리지널 폼타입, 로게인5%폼에어로졸60g](https://i.baropharm.com/products/dc84d96e-d0b4-46bc-bcc8-d62016406fe4.png)
![[레비온] PDRN+EGF, 레비온RX PDRN EGF 크림](https://i.baropharm.com/products/202512/1765949426601.png)
![[한독] 붙이는 통증 전문가, 케토톱 액티브 플라스타(쿨) 40매](https://i.baropharm.com/products/202503/1741829602305.png)
![[알엑스미] 알엑스미 리쥬영 울트라 PDRN 10000 딥리페어 크림](https://i.baropharm.com/partner/products/70c72dd0-cfd3-4d80-87e4-dc4f8de6658b.png?label=바뷰페로고)
![[D판테놀]레비온디판테놀연고](https://i.baropharm.com/products/202607/1782884486469.png)
![[흉터치료]아크리페어겔](https://i.baropharm.com/products/202607/1782884866357.png)
![[여름 한정 특가] 편한가 여름 쿨 세일! (여름 필수템 싹쓰리)](https://gi.esmplus.com/pyunhanga/90.jpg)
![[약물 0%] 터치훅 벌레독소 흡인기](https://cdn-optimized.imweb.me/upload/S2025060969b2a2d28fb0a/7adb5cb72423a.jpg)
![[24H 극강보습] 소이베베 아토 크림](https://cdn-optimized.imweb.me/upload/S2025060969b2a2d28fb0a/c034ac69d4f6c.jpg)
![[올리브베러Pick] 드링킷 건강음료](https://cdn-optimized.imweb.me/upload/S2025060969b2a2d28fb0a/7a29cd38b7da6.jpg)
![[완전방수] 눈시림없는 선크림 (SPF50+)](https://cdn-optimized.imweb.me/upload/S2025060969b2a2d28fb0a/4237159ef592d.png)
![[4.98후기검증] 빛나는 피부 오브링 세럼](https://cdn-optimized.imweb.me/thumbnail/20260520/4db3dcb1296da.jpg)
![[국내최초] 모기디퓨저 천연 계피 모키센트 디퓨저](https://cdn-optimized.imweb.me/upload/S2025060969b2a2d28fb0a/95f4414df691d.jpg)
![[구취 96% 제거] 씹는 고체 가글](https://cdn-optimized.imweb.me/upload/S2025060969b2a2d28fb0a/e97244c42fc7f.jpg)
![[평점 4.9]약사선택 근본 솔루션, 솔티스](https://cdn-optimized.imweb.me/upload/S2025060969b2a2d28fb0a/af01248c58258.jpg)
![[약국BEST!] 뉴비타센스 비타민 흡입기](https://cdn-optimized.imweb.me/upload/S2025060969b2a2d28fb0a/c9b14e1d94ed9.jpg)
![[쿠팡 완판] 수험생 아르기닌 에너지 젤리](https://cdn-optimized.imweb.me/upload/S2025060969b2a2d28fb0a/e4e83872c5505.jpg)
![[100% 천연옥] 멜팅 하트 괄사 마사지기](https://cdn-optimized.imweb.me/upload/S2025060969b2a2d28fb0a/fbf739ccf97a7.jpg)
오늘의 TOP 10
- 1만성질환 '구강붕해정' 상업화 완성 잇따라… 실적도 따라올까
- 2‘4번째 진입 견제’…깐깐한 약가우대의 위험한 담합 유혹
- 3카드 세액공제 연 500만원 축소…매약 매출 높은 약국 부담
- 4경평없이 바로 급여 준다는데, 망설여지는 제도가 있다?
- 5이연제약 트롬빈, 미국 진출 눈앞…고마진 사업 글로벌 확장
- 6의원-약국-유통까지…수면 위로 드러난 '메디컬 플랫폼' 논란
- 7창고형약국 표시광고 규제, 남인순 법안 법사위 통과로 새국면
- 8'소각' 대신 '임직원 보상'…제약바이오 자사주 활용 다변화
- 9"파격혜택 제공" 대범한 창고형 모집공고, 약사들 분통
- 10약사회 "안전상비약 실태 확인부터"…복지부, 점검 검토




