




-
 이연제약 "충주 바이오공장, 파트너 확대 추진"[데일리팜=이석준 기자] 이연제약은 국내 최대규모의 종합 바이오 컨벤션 중 하나인 '바이오플러스-인터펙스코리아 2022(BIX 2022)'에 참가해 파트너십 확대를 진행한다고 3일 밝혔다. 올해로 8 회를 맞는 '바이오플러스-인터펙스코리아'는 한국바이오협회와 리드엑시비션스코리아가 공동 주관하는 컨벤션이다. 국내외 바이오·제약산업의 Value Chain을 공유하고 글로벌 비즈니스 네트워킹을 통한 해외 진출 발판을 마련하는 행사다. 3일부터 사흘간 서울 코엑스에서 열린다. 이연제약은 BIX 2022에 참가해 충주 바이오공장의 cGMP급 생산설비와 생산능력을 선보이며 공동개발 파트너십 확대 및 CDMO/CMO 사업을 논의할 예정이다. 약 800억원이 투입된 충주 바이오공장은 플라스미드 DNA(pDNA), mRNA, 바이러스 벡터, Phage 등 멀티 모달리티(Multi Modality)에 대응하는 바이오의약품 생산공장이다. 원액(Drug Substance, DS)에서 완제(Drug Product, DP)까지 One-Stop으로 생산할 수 있다. 바이오의약품 원액(DS) 생산라인은 다양한 크기의 Multi-Use 및 Single-Use 배양기(Fermenter)를 통해 pDNA 등 미생물 발효기반의 여러 물질들을 동시 다발적으로 생산한다. 임상시료 등 소규모(Small-scale) 생산부터 대규모(Large-Scale) 상용화 생산까지 가능하다. 완제(DP)의 경우 교차오염을 원천적으로 차단하는 Single-Use 생산시설을 통해 미생물 기반 바이오의약품 및 최첨단 항체의약품의 액상 및 동결건조 주사제를 대량으로 생산할 수 있다. 김영민 이연제약 전무이사(바이오사업 총괄)는 "충주 바이오공장은 다양한 파이프라인 단계별 공급망을 구축해 파트너사의 임상개발 기간을 단축할 수 있는 능력을 갖추고 있다. 파트너와 다수 신약 파이프라인 공동 개발을 진행 중이다. BIX 2022 참가를 통해 추가적인 파트너십을 추진하고 있다"고 말했다. 이연제약은 충주공장 생산능력을 바탕으로 CMO/CDMO 사업에도 진행중이다. 올 4월에는 pDNA 4종 공급 계약을 수주했다. 같은 부지에 위치하고 있는 충주 케미칼공장(약 2100억원 투입)도 본격 생산 능력을 갖추기 위한 GMP 인증에 속도를 내고 있다. 케미칼의약품 대규모 생산능력을 기반으로 다수 전통제약사와 CMO 사업을 활발하게 진행하고 있다.2022-08-03 09:40:23이석준
이연제약 "충주 바이오공장, 파트너 확대 추진"[데일리팜=이석준 기자] 이연제약은 국내 최대규모의 종합 바이오 컨벤션 중 하나인 '바이오플러스-인터펙스코리아 2022(BIX 2022)'에 참가해 파트너십 확대를 진행한다고 3일 밝혔다. 올해로 8 회를 맞는 '바이오플러스-인터펙스코리아'는 한국바이오협회와 리드엑시비션스코리아가 공동 주관하는 컨벤션이다. 국내외 바이오·제약산업의 Value Chain을 공유하고 글로벌 비즈니스 네트워킹을 통한 해외 진출 발판을 마련하는 행사다. 3일부터 사흘간 서울 코엑스에서 열린다. 이연제약은 BIX 2022에 참가해 충주 바이오공장의 cGMP급 생산설비와 생산능력을 선보이며 공동개발 파트너십 확대 및 CDMO/CMO 사업을 논의할 예정이다. 약 800억원이 투입된 충주 바이오공장은 플라스미드 DNA(pDNA), mRNA, 바이러스 벡터, Phage 등 멀티 모달리티(Multi Modality)에 대응하는 바이오의약품 생산공장이다. 원액(Drug Substance, DS)에서 완제(Drug Product, DP)까지 One-Stop으로 생산할 수 있다. 바이오의약품 원액(DS) 생산라인은 다양한 크기의 Multi-Use 및 Single-Use 배양기(Fermenter)를 통해 pDNA 등 미생물 발효기반의 여러 물질들을 동시 다발적으로 생산한다. 임상시료 등 소규모(Small-scale) 생산부터 대규모(Large-Scale) 상용화 생산까지 가능하다. 완제(DP)의 경우 교차오염을 원천적으로 차단하는 Single-Use 생산시설을 통해 미생물 기반 바이오의약품 및 최첨단 항체의약품의 액상 및 동결건조 주사제를 대량으로 생산할 수 있다. 김영민 이연제약 전무이사(바이오사업 총괄)는 "충주 바이오공장은 다양한 파이프라인 단계별 공급망을 구축해 파트너사의 임상개발 기간을 단축할 수 있는 능력을 갖추고 있다. 파트너와 다수 신약 파이프라인 공동 개발을 진행 중이다. BIX 2022 참가를 통해 추가적인 파트너십을 추진하고 있다"고 말했다. 이연제약은 충주공장 생산능력을 바탕으로 CMO/CDMO 사업에도 진행중이다. 올 4월에는 pDNA 4종 공급 계약을 수주했다. 같은 부지에 위치하고 있는 충주 케미칼공장(약 2100억원 투입)도 본격 생산 능력을 갖추기 위한 GMP 인증에 속도를 내고 있다. 케미칼의약품 대규모 생산능력을 기반으로 다수 전통제약사와 CMO 사업을 활발하게 진행하고 있다.2022-08-03 09:40:23이석준 -
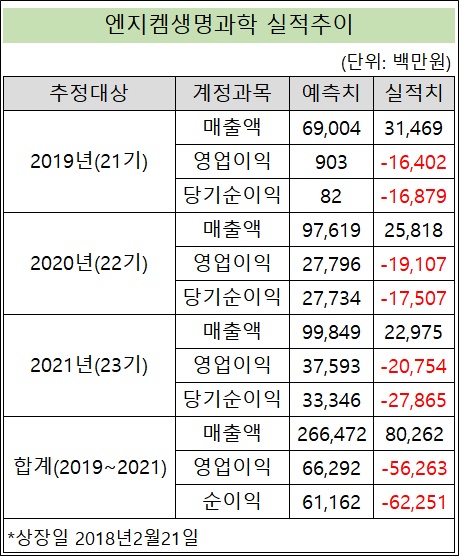 엔지켐, 최근 3년 매출…상장 전 예측치 30% 수준[데일리팜=이석준 기자] 엔지켐생명과학의 최근 3년(2019~2021) 매출액 합계는 상장 전 예측치의 30% 수준에 머무는 것으로 확인됐다. 해당 기간 600억원 이상을 점쳤던 영업이익과 순이익은 되레 600억원 안팎의 손실을 냈다. 엔지켐생명과학은 2일 이같은 내용을 담은 사업보고서 기재정정 공시를 냈다. 회사는 2018년 2월 코넥스에서 코스닥으로 이전 상장했다. 공시를 보면, 엔지켐생명과학의 코스닥 상장을 위한 증권신고서에 기재된 매출액 예측치는 2019년 690억원, 2020년 976억원, 2021년 998억원이다. 3년 합계 2664억원이다. 실상은 달랐다. 실제 매출액은 2019년 315억원, 2020년 258억원, 2021년 230억원으로 합계 803억원이다. 추정치와의 괴리율은 69.86%다. 예측치의 30% 수준에 머물렀다는 소리다. 매출액 괴리율 발생사유는 다양하다. ▲저가형 API 업체(중국, 인도)의 공격적 마케팅 여파로 인한 국내외 영업환경 악화 ▲코로나19로 내수 침체 현상 발생 및 해외 업체 매입 규모 축소 ▲2020년 호중구감소증 2상 Part 2프로토콜 변경 ▲코로나19로 국내외 임상환자 모집 난항으로 호중구감소증 임상과 구강점막염 치료제 2상 및 라이선스 아웃 지연 등이다. 영업이익과 순이익도 예측치와 달랐다. 회사는 2019~2021년 영업이익과 순이익 합계를 각각 663억원, 612억원으로 점쳤다. 다만 실제로는 영업손실 563억원, 순손실 623억원을 기록했다. 해당 기간 매년 영업이익과 순이익 부문은 적자가 지속됐다. 회사는 영업손실 발생 사유에 대해 ▲라이선스 아웃을 통한 신약개발비용의 헷지효과(라이선스아웃으로 인한 임상비용 충당)가 지연되면서 신약개발투자가 경상개발비로 인식됐고 ▲당초 추정자료에 포함되지 않은 신규 파이프라인(COVID-19 치료제 등) 연구개발 비용이 추가로 발생했다고 설명했다. 순손실은 매출액과 영업이익 괴리율 사유에 더해 ▲추정치에 포함되지 않은 전환사채 발행(2020년)에 따른 비현금성 이자비용(전환권조정 상각) 발생(약 103억)이 원인이라고 분석했다. 한편 엔지켐생명과학은 지난달 27일 500%(신주당 5주 발행) 무상증자를 결정했다. 이날 회사는 상한가를 쳤다. 8월 2일 종가는 2만2450원(시총 3128억원)이다. 신주의 배정 기준일은 내달 18일, 상장 예정일은 오는 9월 21일이다.2022-08-03 06:13:02이석준
엔지켐, 최근 3년 매출…상장 전 예측치 30% 수준[데일리팜=이석준 기자] 엔지켐생명과학의 최근 3년(2019~2021) 매출액 합계는 상장 전 예측치의 30% 수준에 머무는 것으로 확인됐다. 해당 기간 600억원 이상을 점쳤던 영업이익과 순이익은 되레 600억원 안팎의 손실을 냈다. 엔지켐생명과학은 2일 이같은 내용을 담은 사업보고서 기재정정 공시를 냈다. 회사는 2018년 2월 코넥스에서 코스닥으로 이전 상장했다. 공시를 보면, 엔지켐생명과학의 코스닥 상장을 위한 증권신고서에 기재된 매출액 예측치는 2019년 690억원, 2020년 976억원, 2021년 998억원이다. 3년 합계 2664억원이다. 실상은 달랐다. 실제 매출액은 2019년 315억원, 2020년 258억원, 2021년 230억원으로 합계 803억원이다. 추정치와의 괴리율은 69.86%다. 예측치의 30% 수준에 머물렀다는 소리다. 매출액 괴리율 발생사유는 다양하다. ▲저가형 API 업체(중국, 인도)의 공격적 마케팅 여파로 인한 국내외 영업환경 악화 ▲코로나19로 내수 침체 현상 발생 및 해외 업체 매입 규모 축소 ▲2020년 호중구감소증 2상 Part 2프로토콜 변경 ▲코로나19로 국내외 임상환자 모집 난항으로 호중구감소증 임상과 구강점막염 치료제 2상 및 라이선스 아웃 지연 등이다. 영업이익과 순이익도 예측치와 달랐다. 회사는 2019~2021년 영업이익과 순이익 합계를 각각 663억원, 612억원으로 점쳤다. 다만 실제로는 영업손실 563억원, 순손실 623억원을 기록했다. 해당 기간 매년 영업이익과 순이익 부문은 적자가 지속됐다. 회사는 영업손실 발생 사유에 대해 ▲라이선스 아웃을 통한 신약개발비용의 헷지효과(라이선스아웃으로 인한 임상비용 충당)가 지연되면서 신약개발투자가 경상개발비로 인식됐고 ▲당초 추정자료에 포함되지 않은 신규 파이프라인(COVID-19 치료제 등) 연구개발 비용이 추가로 발생했다고 설명했다. 순손실은 매출액과 영업이익 괴리율 사유에 더해 ▲추정치에 포함되지 않은 전환사채 발행(2020년)에 따른 비현금성 이자비용(전환권조정 상각) 발생(약 103억)이 원인이라고 분석했다. 한편 엔지켐생명과학은 지난달 27일 500%(신주당 5주 발행) 무상증자를 결정했다. 이날 회사는 상한가를 쳤다. 8월 2일 종가는 2만2450원(시총 3128억원)이다. 신주의 배정 기준일은 내달 18일, 상장 예정일은 오는 9월 21일이다.2022-08-03 06:13:02이석준 -
 신약 '케이캡'의 힘...HK이노엔, 2년새 매출 2배 '껑충'[데일리팜=천승현 기자] HK이노엔이 신약 ‘케이캡’의 높은 성장세를 발판으로 역대 최대 규모 매출을 기록했다. HK이노엔은 지난 2분기 영업이익이 177억원으로 전년동기대비 496.5% 증가했다고 2일 공시했다. 매출액은 2519억원으로 전년보다 36.2% 늘었다. 이 회사의 2분기 매출은 설립 이후 최대 규모다. 지난 2020년 2분기 1309억원에서 2년 만에 2배 가량 성장할 정도로 가파른 성장세를 나태내고 있다. 영업이익은 2020년 4분기 463억원에 이어 역대 두 번째 규모다. 자체개발 신약 케이캡이 성장세를 주도했다. 의약품 조사기관 유비스트에 따르면 케이캡은 지난 2분기 외래 처방금액이 305억원으로 전년보다 19.1% 성장했다. 케이캡은 1분기 처방실적이 301억원으로 전년보다 23.3% 확대된데 이어 2분기에도 성장세를 이어갔다. 케이캡은 상반기에만 606억원의 처방실적을 올렸다. 2019년 3월 발매된 케이캡은 '칼륨 경쟁적 위산분비억제제(P-CAB)’ 계열의 항궤양제다. 위벽 세포에서 산분비 최종 단계에 위치하는 양성자펌프와 칼륨이온을 경쟁적으로 결합시켜 위산 분비를 저해하는 작용기전을 나타낸다. 케이캡은 기존 프로톤펌프억제제(PPI) 계열 제품보다 약효가 빠르게 나타나고, 식사 전후 상관없이 복용이 가능한 점 등 장점을 앞세워 높은 성장세를 지속하고 있다. 케이캡은 발매 첫해 처방금액 309억원을 올리며 돌풍을 일으켰고 지난해에는 출시 3년 차에 처방액 1000억원을 돌파했다. 케이캡은 올해 들어 2월과 4월을 제외하고 모두 월 처방액이 100억원을 넘어섰다. 케이캡의 급여 범위 확대도 가파른 성장세의 요인으로 지목된다. 케이캡은 출시 당시 미란성 위식도역류질환과 비미란성 위식도역류질환 적응증에 건강보험 급여가 적용됐는데 지난해 11월부터 위궤양 영역에도 급여 적용 범위가 확대됐다. 자궁경부암 백신 등 MSD 백신의 주문량이 증가한 점도 매출 성장에 영향을 미쳤다. HK이노엔은 지난해부터 한국MSD의 4가 HPV백신 ‘가다실’ 등 백신 7종의 유통과 판매를 담당하고 있다. 거리두기 해제에 따른 수액제 및 숙취해소제의 매출 증가 효과도 있었다. 병의원 방문 및 수술 건수 증가로 전체 수액 제품군의 수요도 늘었고 모임, 회식 등이 활발해지면서 숙취해소제 컨디션 매출이 지속적으로 증가했다는 게 회사 측 설명이다. HK이노엔은 3, 4분기에도 지속적인 실적 성장을 전망했다. HK이노엔 관계자는 “케이캡은 추가 글로벌 진출 계약 및 미국 후속 임상을준비 중으로,국내 및 글로벌 시장에서의 활약이 계속될 것”이라며 “숙취해소제는 컨디션스틱을 필두로 MZ세대까지 소비층을 지속 넓힐 예정으로 시장을 선도하는 핵심제품들로 내실있는 성장이 이어지도록 할 것”이라고 말했다.2022-08-02 12:11:11천승현
신약 '케이캡'의 힘...HK이노엔, 2년새 매출 2배 '껑충'[데일리팜=천승현 기자] HK이노엔이 신약 ‘케이캡’의 높은 성장세를 발판으로 역대 최대 규모 매출을 기록했다. HK이노엔은 지난 2분기 영업이익이 177억원으로 전년동기대비 496.5% 증가했다고 2일 공시했다. 매출액은 2519억원으로 전년보다 36.2% 늘었다. 이 회사의 2분기 매출은 설립 이후 최대 규모다. 지난 2020년 2분기 1309억원에서 2년 만에 2배 가량 성장할 정도로 가파른 성장세를 나태내고 있다. 영업이익은 2020년 4분기 463억원에 이어 역대 두 번째 규모다. 자체개발 신약 케이캡이 성장세를 주도했다. 의약품 조사기관 유비스트에 따르면 케이캡은 지난 2분기 외래 처방금액이 305억원으로 전년보다 19.1% 성장했다. 케이캡은 1분기 처방실적이 301억원으로 전년보다 23.3% 확대된데 이어 2분기에도 성장세를 이어갔다. 케이캡은 상반기에만 606억원의 처방실적을 올렸다. 2019년 3월 발매된 케이캡은 '칼륨 경쟁적 위산분비억제제(P-CAB)’ 계열의 항궤양제다. 위벽 세포에서 산분비 최종 단계에 위치하는 양성자펌프와 칼륨이온을 경쟁적으로 결합시켜 위산 분비를 저해하는 작용기전을 나타낸다. 케이캡은 기존 프로톤펌프억제제(PPI) 계열 제품보다 약효가 빠르게 나타나고, 식사 전후 상관없이 복용이 가능한 점 등 장점을 앞세워 높은 성장세를 지속하고 있다. 케이캡은 발매 첫해 처방금액 309억원을 올리며 돌풍을 일으켰고 지난해에는 출시 3년 차에 처방액 1000억원을 돌파했다. 케이캡은 올해 들어 2월과 4월을 제외하고 모두 월 처방액이 100억원을 넘어섰다. 케이캡의 급여 범위 확대도 가파른 성장세의 요인으로 지목된다. 케이캡은 출시 당시 미란성 위식도역류질환과 비미란성 위식도역류질환 적응증에 건강보험 급여가 적용됐는데 지난해 11월부터 위궤양 영역에도 급여 적용 범위가 확대됐다. 자궁경부암 백신 등 MSD 백신의 주문량이 증가한 점도 매출 성장에 영향을 미쳤다. HK이노엔은 지난해부터 한국MSD의 4가 HPV백신 ‘가다실’ 등 백신 7종의 유통과 판매를 담당하고 있다. 거리두기 해제에 따른 수액제 및 숙취해소제의 매출 증가 효과도 있었다. 병의원 방문 및 수술 건수 증가로 전체 수액 제품군의 수요도 늘었고 모임, 회식 등이 활발해지면서 숙취해소제 컨디션 매출이 지속적으로 증가했다는 게 회사 측 설명이다. HK이노엔은 3, 4분기에도 지속적인 실적 성장을 전망했다. HK이노엔 관계자는 “케이캡은 추가 글로벌 진출 계약 및 미국 후속 임상을준비 중으로,국내 및 글로벌 시장에서의 활약이 계속될 것”이라며 “숙취해소제는 컨디션스틱을 필두로 MZ세대까지 소비층을 지속 넓힐 예정으로 시장을 선도하는 핵심제품들로 내실있는 성장이 이어지도록 할 것”이라고 말했다.2022-08-02 12:11:11천승현 -
 K-제약바이오 생산기지 경쟁력 쑥…FDA 잇단 눈도장[데일리팜=이석준 기자] K-제약바이오 생산기지 경쟁력이 올라가고 있다. 가장 까다롭다고 평가받는 미국 FDA(식품의약국) 실사 통과 사례가 늘고 있어서다. 해당 기업은 글로벌 진출을 본격화한다는 방침이다. 아시아 최초 올리고 제조소 인증 에스티팜은 얼마전 아시아 최초 올리고 제조소로 FDA cGMP(우수의약품품질관리기준) 인증을 획득했다. 반월 캠퍼스 올리고동 PAI(신약 승인 전 제조소 실사) 결과 무결점(NAI) 등급을 받으며 시설 R&D 역량을 입증했다. 올리고 수주 확대가 예고된다. 에스티팜은 지금까지 미국 시장에 임상용 올리고핵산치료제 원료의약품 수출만 가능했다. 이번 FDA cGMP 승인으로 미국에 대규모 상업화 물량 수출까지 가능해졌다. 회사 관계자는 "내년 상반기까지 4건의 FDA PAI실사가 예정됐다. 이는 에스티팜 원료 공급 신약들의 FDA 승인이 임박했음을 의미한다. 이번 FDA cGMP 승인은 향후 진행될 실사에도 긍정적인 영향을 미칠 것으로 예상된다"고 말했다. 국내 첫 항암제 API 제조소 등록 이니스트에스티 오송공장도 최근 항암제 API전용 cGMP 제조소로 FDA에 등록됐다. 이에 오송공장은 고활성 비세포독성 항암제 API전용 제조소로 국내 최초 cGMP 인증 항암제 원료의약품 제조시설이 됐다. 회사 관계자는 "FDA 실사 통과가 알려지면서 미국 등 선진 의약품 시장에 진출하고자 하는 기존 및 신규 거래처로부터 러브콜이 이어지고 있다. 글로벌 수준 API 제조설비와 품질관리 능력을 재입증했다"고 강조했다. FDA 실사 통과로 이니스트에스티 선투자도 탄력이 받게 됐다. 회사는 지난해 10월 CMO(위탁생산) 비지니즈 확대를 위해 오송공장 부지에 400억원 규모의 '글로벌 API합성 cGMP 플랜트'를 착공했다. 오는 10월 준공을 앞두고 있다. 코로나 mRNA 백신 완제 이어 원료 생산 성공 삼성바이오로직스는 코로나19 mRNA 백신 원료의약품 시험생산에 성공했다. 지난해 하반기 모더나 mRNA 백신 완제의약품 위탁생산(CMO)에 이어 mRNA 원료의약품 설비까지 마련하며 원스톱 생산체제를 갖추게 됐다. 회사는 지난해말 미국 그린라이트가 개발중인 코로나19 백신 후보물질의 원료의약품(DS) 위탁생산 파트너십을 체결했다. 이후 7개월간 기술이전 및 스케일업을 거쳐 지난 5월말 mRNA 원료의약품 생산설비 구축을 완료했다. 삼성바이오로직스는 최근 첫 시험생산에 성공하며 mRNA 백신 원료의약품을 대규모 상업생산 할 수 있는 준비를 마쳤다. 시험생산은 본격적인 상업 생산 직전에 실시하는 공정 검증 단계다. 시험생산 성공은 cGMP 수준의 생산 능력을 갖췄다고 평가받는다. 두번째 시험 생산은 이달 중 시행될 예정이다. 시장 관계자는 "FDA 실사 통과 등 차별화된 생산기지를 갖춘 제약바이오 기업들이 늘고 있다. 시설 R&D도 물질 R&D 만큼 기업 경쟁력으로 취급받는 시대가 왔다"고 진단했다.2022-08-02 12:00:38이석준
K-제약바이오 생산기지 경쟁력 쑥…FDA 잇단 눈도장[데일리팜=이석준 기자] K-제약바이오 생산기지 경쟁력이 올라가고 있다. 가장 까다롭다고 평가받는 미국 FDA(식품의약국) 실사 통과 사례가 늘고 있어서다. 해당 기업은 글로벌 진출을 본격화한다는 방침이다. 아시아 최초 올리고 제조소 인증 에스티팜은 얼마전 아시아 최초 올리고 제조소로 FDA cGMP(우수의약품품질관리기준) 인증을 획득했다. 반월 캠퍼스 올리고동 PAI(신약 승인 전 제조소 실사) 결과 무결점(NAI) 등급을 받으며 시설 R&D 역량을 입증했다. 올리고 수주 확대가 예고된다. 에스티팜은 지금까지 미국 시장에 임상용 올리고핵산치료제 원료의약품 수출만 가능했다. 이번 FDA cGMP 승인으로 미국에 대규모 상업화 물량 수출까지 가능해졌다. 회사 관계자는 "내년 상반기까지 4건의 FDA PAI실사가 예정됐다. 이는 에스티팜 원료 공급 신약들의 FDA 승인이 임박했음을 의미한다. 이번 FDA cGMP 승인은 향후 진행될 실사에도 긍정적인 영향을 미칠 것으로 예상된다"고 말했다. 국내 첫 항암제 API 제조소 등록 이니스트에스티 오송공장도 최근 항암제 API전용 cGMP 제조소로 FDA에 등록됐다. 이에 오송공장은 고활성 비세포독성 항암제 API전용 제조소로 국내 최초 cGMP 인증 항암제 원료의약품 제조시설이 됐다. 회사 관계자는 "FDA 실사 통과가 알려지면서 미국 등 선진 의약품 시장에 진출하고자 하는 기존 및 신규 거래처로부터 러브콜이 이어지고 있다. 글로벌 수준 API 제조설비와 품질관리 능력을 재입증했다"고 강조했다. FDA 실사 통과로 이니스트에스티 선투자도 탄력이 받게 됐다. 회사는 지난해 10월 CMO(위탁생산) 비지니즈 확대를 위해 오송공장 부지에 400억원 규모의 '글로벌 API합성 cGMP 플랜트'를 착공했다. 오는 10월 준공을 앞두고 있다. 코로나 mRNA 백신 완제 이어 원료 생산 성공 삼성바이오로직스는 코로나19 mRNA 백신 원료의약품 시험생산에 성공했다. 지난해 하반기 모더나 mRNA 백신 완제의약품 위탁생산(CMO)에 이어 mRNA 원료의약품 설비까지 마련하며 원스톱 생산체제를 갖추게 됐다. 회사는 지난해말 미국 그린라이트가 개발중인 코로나19 백신 후보물질의 원료의약품(DS) 위탁생산 파트너십을 체결했다. 이후 7개월간 기술이전 및 스케일업을 거쳐 지난 5월말 mRNA 원료의약품 생산설비 구축을 완료했다. 삼성바이오로직스는 최근 첫 시험생산에 성공하며 mRNA 백신 원료의약품을 대규모 상업생산 할 수 있는 준비를 마쳤다. 시험생산은 본격적인 상업 생산 직전에 실시하는 공정 검증 단계다. 시험생산 성공은 cGMP 수준의 생산 능력을 갖췄다고 평가받는다. 두번째 시험 생산은 이달 중 시행될 예정이다. 시장 관계자는 "FDA 실사 통과 등 차별화된 생산기지를 갖춘 제약바이오 기업들이 늘고 있다. 시설 R&D도 물질 R&D 만큼 기업 경쟁력으로 취급받는 시대가 왔다"고 진단했다.2022-08-02 12:00:38이석준 -
 삼진제약, 캐나다 AI 업체와 신약 개발 나선다[데일리팜=이석준 기자] 삼진제약은 캐나다 인공지능 신약개발 플랫폼 기업 '사이클리카(Cyclica CEO)와 AI 신약개발 공동 연구 계약을 체결했다고 2일 밝혔다. 협약을 통해 삼진제약은 현재 검토 중에 있는 복수의 약물 타겟을 사이클리카에 제안하게 된다. 사이클리카는 AI기반 신약 후보물질 발굴 플랫폼(Ligand DesignTM) 기술을 적용해 개발 가능성 높은 후보 물질을 확보한다. 이수민 삼진제약 연구센터장은 "사이클리카와의 AI 기술을 활용한 공동 연구로 신약 개발에 소요되는 시간과 비용을 현저히 줄일 수 있게 됐다. 오픈이노베이션 전략을 통해 혁신 신약들을 효율적으로 발굴하고 개발할 수 있도록 국내외 유수의 연구기관 및 기업들과 협력할 것"이라고 말했다. 사이클리카는 2020년 기술 시장 조사기관 'CB Insights'로부터 세계 13대 헬스케어 AI 스타트업 중 하나로 선정됐다. Merck KGaA, AstraZeneca 등 글로벌 빅파마를 포함한 국내외 다수 회사와 공동 연구를 진행중이다.2022-08-02 08:49:14이석준
삼진제약, 캐나다 AI 업체와 신약 개발 나선다[데일리팜=이석준 기자] 삼진제약은 캐나다 인공지능 신약개발 플랫폼 기업 '사이클리카(Cyclica CEO)와 AI 신약개발 공동 연구 계약을 체결했다고 2일 밝혔다. 협약을 통해 삼진제약은 현재 검토 중에 있는 복수의 약물 타겟을 사이클리카에 제안하게 된다. 사이클리카는 AI기반 신약 후보물질 발굴 플랫폼(Ligand DesignTM) 기술을 적용해 개발 가능성 높은 후보 물질을 확보한다. 이수민 삼진제약 연구센터장은 "사이클리카와의 AI 기술을 활용한 공동 연구로 신약 개발에 소요되는 시간과 비용을 현저히 줄일 수 있게 됐다. 오픈이노베이션 전략을 통해 혁신 신약들을 효율적으로 발굴하고 개발할 수 있도록 국내외 유수의 연구기관 및 기업들과 협력할 것"이라고 말했다. 사이클리카는 2020년 기술 시장 조사기관 'CB Insights'로부터 세계 13대 헬스케어 AI 스타트업 중 하나로 선정됐다. Merck KGaA, AstraZeneca 등 글로벌 빅파마를 포함한 국내외 다수 회사와 공동 연구를 진행중이다.2022-08-02 08:49:14이석준 -
 '든든한 캐시카우 덕분에'...제약사들, 매출 신기록 행진[데일리팜=천승현 기자] 대형 제약바이오기업들이 대내외적으로 불안정한 환경에도 지난 2분기 준수한 성적표를 받아들었다. 위탁사업, 처방의약품, 일반의약품, 백신 등 주력 사업들이 안정적인 성장을 견인하며 매출 신기록을 쏟아냈다. 1일 금융감독원에 따르면 주요 제약바이오기업 10곳 중 9곳의 매출이 전년동기대비 확대됐다. 삼성바이오로직스, 유한양행, 녹십자, 종근당, 한미약품, 대웅제약, 보령제약, 일동제약, 동아에스티, SK바이오사이언스 잠정 실적을 발표한 주요 제약바이오기업 10곳을 대상으로 집계했다. 이중 SK바이오사이언스만 매출 규모가 전년대비 줄었다. 삼성바이오로직스, 유한양행, 종근당, 대웅제약, 보령제약, 일동제약 등은 분기 매출 신기록을 갈아치웠다. 주요 제약바이오기업 10곳 중 5곳은 영업이익이 작년보다 개선됐다. ◆삼바, 국내 제약바이오 첫 반기 매출 1조 돌파...유한, 내수·해외시장 동반 호조 삼성바이오로직스가 가장 눈에 띄는 실적을 냈다. 2분기 매출이 6514억원으로 전년보다 58.1% 늘었고 영업이익은 1.8% 증가한 1697억원을 기록했다. 매출은 창립 이후 최대 규모다. 삼성바이오로직스는 바이오 의약품 수탁 생산(CMO)이 주력 사업이다. 지난 2018년 10월 단일 공장으로는 세계 최대 규모(18만리터) 3공장이 본격적으로 가동하면서 위탁 계약 물량도 급증하고 있다. 삼성바이오로직스는 상반기 기준 누적 CMO 수주 73건을 기록했다. 올해 상반기에는 얀센, 머크, GSK, 일라이 릴리, 노바티스 등 대형 제약사들과 계약을 잇따라 체결하며 누적 수주 총액 79억 달러를 달성했다. 삼성바이오로직스는 상반기 누적 매출 1조1617억원으로 전년보다 72.8% 치솟았다. 삼성바이오로직스는 의약품을 취급하는 국내 제약바이오기업 중 처음을 반기 매출 1조원을 돌파했다. 유한양행은 내수와 해외 시장에서 주력사업의 동반 성장으로 분기 매출 신기록을 달성했다. 유한양행은 2분기 매출액이 4680억원으로 전년보다 10.4% 늘었다. 지난 2020년 4분기에 올린 4394억원을 6분기만에 넘어섰다. 내수 시장에서는 비처방약(OTC) 사업의 매출이 491억원으로 전년동기 397억원보다 23.9% 확대됐다. 2분기 처방약(ETC) 매출은 2836억원으로 전년대비 8.9% 늘었다. 감기약 코푸의 매출이 작년 2분기 29억원에서 73억원으로 154.2% 증가했다. 코로나19 확산에 따른 감기약 등의 수요 증가로 반짝 수혜를 입었다. 유한양행의 2분기 해외사업 매출은 568억원으로 작년 같은 기간 366억원보다 55.1% 뛰었다. 유한양행은 유한화학이 생산하는 원료의약품을 사들여 다국적제약사에 수출한다. 다만 기술료 수익 감소와 R&D 투자 증가로 수익성은 악화했다. 유한양행의 2분기 기술료 수익은 52억원으로 전년동기 167억원보다 69.0% 축소됐다. R&D비용은 378억원으로 작년보다 15.8% 확대됐다. ◆녹십자·종근당, 전 사업부 호조 매출 '껑충' 녹십자는 전 사업부가 성장하며 실적 호조를 나타냈다. 녹십자는 지난 2분기 영업이익이 131억원으로 전년동기대비 18.0% 늘었고 매출 4232억원으로 전년보다 9.2% 증가했다. 2분기 매출은 작년 3분기의 4657억원에 이어 역대 두 번째 규모다. 2분기 매출만 보면 창립 이후 최대 규모다. 미국에서 개발 중인 차세대 대상포진 백신 임상 2상의 영향으로 연구개발비가 전년 대비 대폭 증가했지만 주력 제품 호실적을 통해 수익성은 개선됐다. 혈액제제 매출이 15% 증가한 1060억원을 기록했고 백신 매출은 844억원으로 전년대비 8% 확대됐다. 처방의약품 매출은 811억원으로 전년대비 5% 늘었고 소비자헬스케어 등 기타 부문이 지난해보다 8% 성장한 509억원을 나타냈다. 종근당은 처방의약품의 견조한 성장을 기반으로 분기 매출 신기록을 세웠다. 종근당은 지난 2분기 영업이익이 281억원으로 전년 동기 대비 16.5% 감소했지만 매출은 3548억원으로 전년보다 11.6% 늘었다. 2020년 3분기 기록한 매출 신기록을 3575억원을 7분기 만에 경신했다. 주력 처방의약품 모두 선전했다. 뇌기능개선제 종근당글리아티린은 2분기 외래 처방금액이 237억원으로 전년보다 6.2% 증가했다. 골관절염치료제 이모튼은 2분기 처방액이 133억원으로 전년 대비 2.9% 증가했다. 고혈압복합제 텔미누보는 전년 동기 115억원보다 9.3% 증가한 126억원의 처방실적을 기록했다. 최근에는 진단키트와 감기약 판매도 매출 성장에 기여했다. 종근당은 휴마시스와 손 잡고 코로나19 항원 진단키트를 공급하고 있다. 또 코로나19 확진자 급증으로 일반의약품 감기약 모드시리즈 판매도 크게 늘었다. ◆한미, 자체개발 복합제 성장 견인...대웅, 보툴리눔제제 수출 급증 한미약품은 자체개발 복합제를 앞세워 매출과 영업이익 모두 껑충 뛰었다. 한미약품의 2분기 영업이익은 296억원으로 전년 동기보다 86.4% 증가했고 매출액은 3165억원으로 13.3% 늘었다. 고지혈증복합제 로수젯은 상반기 처방액이 686억원으로 전년 동기 대비 13.3% 늘었다. 2015년 말 출시된 로수젯은 로수바스타틴과 에제티미브 2개 성분으로 구성된 복합제다. 로수젯은 2016년 243억원 처방실적을 기록한 이후 매년 가파른 성장세를 나타내고 있다. 2020년과 지난해 2년 연속 처방액 1000억원을 넘어섰다. 한미약품의 간판 복합신약 아모잘탄패밀리도 안정적인 성장세를 지속했다. 한미약품은 암로디핀과 로사르탄 성분이 결합된 복합제 아모잘탄과 함께 아모잘탄플러스, 아모잘탄큐, 아모잘탄엑스큐 등을 판매 중이다. 항생제, 해열진통제 등 코로나19 증상 완화 치료제로 사용되는 제품들도 처방 규모가 큰 폭으로 늘었다. 지난해부터 가파른 성장세를 보이는 북경한미약품이 호실적을 내면서 모기업의 견고한 실적에 기여했다. 대웅제약은 보툴리눔독소제제, 전문의약품, 일반의약품 등의 고른 성장으로 분기 매출 신기록을 새롭게 작성했다. 대웅제약의 2분기 매출은 전년동기대비 7.6% 증가한 2938억원을 나타냈고 영업이익은 336억원으로 25.8% 늘었다. 매출과 영업이익 모두 역대 최대 규모다. 보툴리눔독소제제 나보타의 2분기 매출은 371억원으로 전년 동기 대비 60.0% 늘었다. 전 분기 304억원보다 22.0% 증가하며 역대 최대 규모를 기록했다. 수출 실적 성장세가 돋보였다. 2분기 나보타의 수출액은 292억원으로 전년보다 2배 이상 치솟았다. 1분기에 기록한 종전 신기록 228억원을 크게 넘어섰다. 2분기 나보타의 매출에서 수출이 차지하는 비중은 78.7%에 달했다. 대웅제약은 전문의약품과 일반의약품 매출이 전년보다 각각 5.5%, 19.9% 증가하며 실적 상승을 이끌었다. ◆보령·일동, 주력사업 성장으로 매출 신기록 보령과 일동제약도 분기 매출 신기록을 갈아치웠다. 보령은 지난 2분기 영업이익이 매출은 1722억원으로 전년보다 21.4% 늘었다. 지난해 3분기에 1583억원으로 매출 기록을 세운 이후 4분기 연속 신기록 행진을 이어가고 있다. 보령의 간판 의약품 카나브패밀리는 2분기 매출 326억원으로 전년대비 21% 성장했다. 보령이 자체개발 고혈압신약 카나브에 또 다른 의약품을 결합한 복합제를 속속 내놓고 시장을 공략하고 있다. 보령이 판매 중인 카나브 기반 의약품은 총 6종이다. 항암제 사업은 전년 대비 60% 성장한 364억원의 매출을 기록했다. 수탁 사업도 경제활동 재개 효과로 정상화 되면서 2분기 매출 156억원으로 전년대비 81% 성장했다. 일동제약은 2분기 매출이 전년대비 14.6% 증가한 1620억원을 기록하며 역대 최대 규모를 나타냈다. 일동제약은 올해부터 아스트라제네카와 손 잡고 판매를 시작한 넥시움이 매출 증대 효과가 발생했다. 일동제약이 레피젠과 협업을 통해 신속항원검사키트도 매출 확대에 기여했다. 다만 일동제약은 R&D 투자 확대로 영업손실 220억원을 나타냈다. 일동제약은 2020년 4분기 59억원의 영업손실을 낸 이후 7분기 연속 적자를 기록 중이다. 이 기간 적자 규모는 총 916억원에 달했다. 일동제약은 2분기에만 R&D 비용으로 역대 최대 규모인 341억원을 투자했다. 전년동기보다 16.8% 늘었고 2년 전과 비교하면 2020년 2분기 116억원에서 2년새 3배 가량 확대됐다.2022-08-02 06:20:36천승현
'든든한 캐시카우 덕분에'...제약사들, 매출 신기록 행진[데일리팜=천승현 기자] 대형 제약바이오기업들이 대내외적으로 불안정한 환경에도 지난 2분기 준수한 성적표를 받아들었다. 위탁사업, 처방의약품, 일반의약품, 백신 등 주력 사업들이 안정적인 성장을 견인하며 매출 신기록을 쏟아냈다. 1일 금융감독원에 따르면 주요 제약바이오기업 10곳 중 9곳의 매출이 전년동기대비 확대됐다. 삼성바이오로직스, 유한양행, 녹십자, 종근당, 한미약품, 대웅제약, 보령제약, 일동제약, 동아에스티, SK바이오사이언스 잠정 실적을 발표한 주요 제약바이오기업 10곳을 대상으로 집계했다. 이중 SK바이오사이언스만 매출 규모가 전년대비 줄었다. 삼성바이오로직스, 유한양행, 종근당, 대웅제약, 보령제약, 일동제약 등은 분기 매출 신기록을 갈아치웠다. 주요 제약바이오기업 10곳 중 5곳은 영업이익이 작년보다 개선됐다. ◆삼바, 국내 제약바이오 첫 반기 매출 1조 돌파...유한, 내수·해외시장 동반 호조 삼성바이오로직스가 가장 눈에 띄는 실적을 냈다. 2분기 매출이 6514억원으로 전년보다 58.1% 늘었고 영업이익은 1.8% 증가한 1697억원을 기록했다. 매출은 창립 이후 최대 규모다. 삼성바이오로직스는 바이오 의약품 수탁 생산(CMO)이 주력 사업이다. 지난 2018년 10월 단일 공장으로는 세계 최대 규모(18만리터) 3공장이 본격적으로 가동하면서 위탁 계약 물량도 급증하고 있다. 삼성바이오로직스는 상반기 기준 누적 CMO 수주 73건을 기록했다. 올해 상반기에는 얀센, 머크, GSK, 일라이 릴리, 노바티스 등 대형 제약사들과 계약을 잇따라 체결하며 누적 수주 총액 79억 달러를 달성했다. 삼성바이오로직스는 상반기 누적 매출 1조1617억원으로 전년보다 72.8% 치솟았다. 삼성바이오로직스는 의약품을 취급하는 국내 제약바이오기업 중 처음을 반기 매출 1조원을 돌파했다. 유한양행은 내수와 해외 시장에서 주력사업의 동반 성장으로 분기 매출 신기록을 달성했다. 유한양행은 2분기 매출액이 4680억원으로 전년보다 10.4% 늘었다. 지난 2020년 4분기에 올린 4394억원을 6분기만에 넘어섰다. 내수 시장에서는 비처방약(OTC) 사업의 매출이 491억원으로 전년동기 397억원보다 23.9% 확대됐다. 2분기 처방약(ETC) 매출은 2836억원으로 전년대비 8.9% 늘었다. 감기약 코푸의 매출이 작년 2분기 29억원에서 73억원으로 154.2% 증가했다. 코로나19 확산에 따른 감기약 등의 수요 증가로 반짝 수혜를 입었다. 유한양행의 2분기 해외사업 매출은 568억원으로 작년 같은 기간 366억원보다 55.1% 뛰었다. 유한양행은 유한화학이 생산하는 원료의약품을 사들여 다국적제약사에 수출한다. 다만 기술료 수익 감소와 R&D 투자 증가로 수익성은 악화했다. 유한양행의 2분기 기술료 수익은 52억원으로 전년동기 167억원보다 69.0% 축소됐다. R&D비용은 378억원으로 작년보다 15.8% 확대됐다. ◆녹십자·종근당, 전 사업부 호조 매출 '껑충' 녹십자는 전 사업부가 성장하며 실적 호조를 나타냈다. 녹십자는 지난 2분기 영업이익이 131억원으로 전년동기대비 18.0% 늘었고 매출 4232억원으로 전년보다 9.2% 증가했다. 2분기 매출은 작년 3분기의 4657억원에 이어 역대 두 번째 규모다. 2분기 매출만 보면 창립 이후 최대 규모다. 미국에서 개발 중인 차세대 대상포진 백신 임상 2상의 영향으로 연구개발비가 전년 대비 대폭 증가했지만 주력 제품 호실적을 통해 수익성은 개선됐다. 혈액제제 매출이 15% 증가한 1060억원을 기록했고 백신 매출은 844억원으로 전년대비 8% 확대됐다. 처방의약품 매출은 811억원으로 전년대비 5% 늘었고 소비자헬스케어 등 기타 부문이 지난해보다 8% 성장한 509억원을 나타냈다. 종근당은 처방의약품의 견조한 성장을 기반으로 분기 매출 신기록을 세웠다. 종근당은 지난 2분기 영업이익이 281억원으로 전년 동기 대비 16.5% 감소했지만 매출은 3548억원으로 전년보다 11.6% 늘었다. 2020년 3분기 기록한 매출 신기록을 3575억원을 7분기 만에 경신했다. 주력 처방의약품 모두 선전했다. 뇌기능개선제 종근당글리아티린은 2분기 외래 처방금액이 237억원으로 전년보다 6.2% 증가했다. 골관절염치료제 이모튼은 2분기 처방액이 133억원으로 전년 대비 2.9% 증가했다. 고혈압복합제 텔미누보는 전년 동기 115억원보다 9.3% 증가한 126억원의 처방실적을 기록했다. 최근에는 진단키트와 감기약 판매도 매출 성장에 기여했다. 종근당은 휴마시스와 손 잡고 코로나19 항원 진단키트를 공급하고 있다. 또 코로나19 확진자 급증으로 일반의약품 감기약 모드시리즈 판매도 크게 늘었다. ◆한미, 자체개발 복합제 성장 견인...대웅, 보툴리눔제제 수출 급증 한미약품은 자체개발 복합제를 앞세워 매출과 영업이익 모두 껑충 뛰었다. 한미약품의 2분기 영업이익은 296억원으로 전년 동기보다 86.4% 증가했고 매출액은 3165억원으로 13.3% 늘었다. 고지혈증복합제 로수젯은 상반기 처방액이 686억원으로 전년 동기 대비 13.3% 늘었다. 2015년 말 출시된 로수젯은 로수바스타틴과 에제티미브 2개 성분으로 구성된 복합제다. 로수젯은 2016년 243억원 처방실적을 기록한 이후 매년 가파른 성장세를 나타내고 있다. 2020년과 지난해 2년 연속 처방액 1000억원을 넘어섰다. 한미약품의 간판 복합신약 아모잘탄패밀리도 안정적인 성장세를 지속했다. 한미약품은 암로디핀과 로사르탄 성분이 결합된 복합제 아모잘탄과 함께 아모잘탄플러스, 아모잘탄큐, 아모잘탄엑스큐 등을 판매 중이다. 항생제, 해열진통제 등 코로나19 증상 완화 치료제로 사용되는 제품들도 처방 규모가 큰 폭으로 늘었다. 지난해부터 가파른 성장세를 보이는 북경한미약품이 호실적을 내면서 모기업의 견고한 실적에 기여했다. 대웅제약은 보툴리눔독소제제, 전문의약품, 일반의약품 등의 고른 성장으로 분기 매출 신기록을 새롭게 작성했다. 대웅제약의 2분기 매출은 전년동기대비 7.6% 증가한 2938억원을 나타냈고 영업이익은 336억원으로 25.8% 늘었다. 매출과 영업이익 모두 역대 최대 규모다. 보툴리눔독소제제 나보타의 2분기 매출은 371억원으로 전년 동기 대비 60.0% 늘었다. 전 분기 304억원보다 22.0% 증가하며 역대 최대 규모를 기록했다. 수출 실적 성장세가 돋보였다. 2분기 나보타의 수출액은 292억원으로 전년보다 2배 이상 치솟았다. 1분기에 기록한 종전 신기록 228억원을 크게 넘어섰다. 2분기 나보타의 매출에서 수출이 차지하는 비중은 78.7%에 달했다. 대웅제약은 전문의약품과 일반의약품 매출이 전년보다 각각 5.5%, 19.9% 증가하며 실적 상승을 이끌었다. ◆보령·일동, 주력사업 성장으로 매출 신기록 보령과 일동제약도 분기 매출 신기록을 갈아치웠다. 보령은 지난 2분기 영업이익이 매출은 1722억원으로 전년보다 21.4% 늘었다. 지난해 3분기에 1583억원으로 매출 기록을 세운 이후 4분기 연속 신기록 행진을 이어가고 있다. 보령의 간판 의약품 카나브패밀리는 2분기 매출 326억원으로 전년대비 21% 성장했다. 보령이 자체개발 고혈압신약 카나브에 또 다른 의약품을 결합한 복합제를 속속 내놓고 시장을 공략하고 있다. 보령이 판매 중인 카나브 기반 의약품은 총 6종이다. 항암제 사업은 전년 대비 60% 성장한 364억원의 매출을 기록했다. 수탁 사업도 경제활동 재개 효과로 정상화 되면서 2분기 매출 156억원으로 전년대비 81% 성장했다. 일동제약은 2분기 매출이 전년대비 14.6% 증가한 1620억원을 기록하며 역대 최대 규모를 나타냈다. 일동제약은 올해부터 아스트라제네카와 손 잡고 판매를 시작한 넥시움이 매출 증대 효과가 발생했다. 일동제약이 레피젠과 협업을 통해 신속항원검사키트도 매출 확대에 기여했다. 다만 일동제약은 R&D 투자 확대로 영업손실 220억원을 나타냈다. 일동제약은 2020년 4분기 59억원의 영업손실을 낸 이후 7분기 연속 적자를 기록 중이다. 이 기간 적자 규모는 총 916억원에 달했다. 일동제약은 2분기에만 R&D 비용으로 역대 최대 규모인 341억원을 투자했다. 전년동기보다 16.8% 늘었고 2년 전과 비교하면 2020년 2분기 116억원에서 2년새 3배 가량 확대됐다.2022-08-02 06:20:36천승현 -
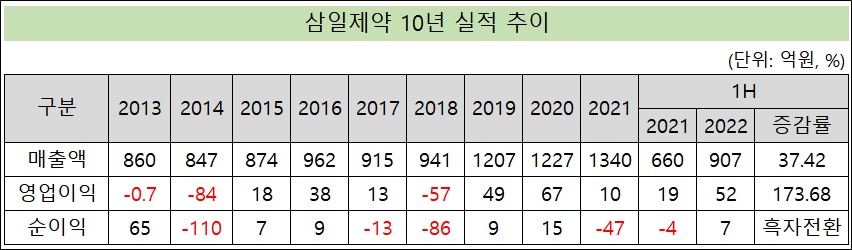 호실적 삼일제약, 단숨에 연 매출 2천억 돌파 가시권[데일리팜=이석준 기자] 반기 호실적을 낸 삼일제약이 연매출 2000억원 돌파를 가시권에 뒀다. 기존 최대 매출은 지난해 1340억원이다. 영업이익은 2008년(108억원) 이후 첫 100억원을 넘어설 기세다. 부루펜(OTC), ETC, 안과, CNS(중추신경계) 등 전 사업부가 고른 성장을 거둔 덕분이다. 방점은 올해 9월 완공이 예정된 베트남 점안제 공장이다. 공장 본격 가동시 CMO(위탁생산) 사업 확대로 이어진다. 삼일제약 잠정 공시에 따르면, 회사의 개별 기준 올 반기 매출액과 영업이익은 각각 907억원, 52억원이다. 지난해 상반기와 견줘 37.42%, 173.68% 늘었다. 창립 최대 실적이 점쳐진다. 단순 계산시 연간 매출액은 1814억원, 영업이익 104억원이다. 상승세를 감안하면 외형은 첫 2000억원 돌파도 가능해보인다. 매출 2000억원은 기존 최대인 지난해 1340억원보다 50% 가까이 늘은 수치다. 영업이익은 2008년(108억원) 이후 첫 100억원을 넘기게 된다. 반기 호실적은 '포리부틴', '리박트과립' 등 ETC 주요 품목과 안과, CNS, 위수탁 사업부 등 전 사업부의 고른 성장에 힘입은 결과다. 코로나 여파로 어린이해열제 '어린이부루펜시럽' 판매량도 급증했다. 상반기 매출만 39억원 규모로 전년(28억원)을 넘어섰다. 회사 관계자는 "안정적 재고 확보를 위해 월평균 판매량 이상 재고를 확보하도록 노력하고 있다. 전사 휴가 중에도 어린이부루펜시럽 생산라인의 일부 인원이 근무하여 재고 확보를 위해 총력을 기하고 있다"고 말했다. 잇단 유통 계약…9월 준공 베트남 공장 방점 신규 라인업 보강으로 추가 동력도 마련했다. 회사는 최근 삼성바이오에피스와 황반변성, 당뇨병성 황반부종 치료제 '아멜리부' 국내 독점 유통 판매를 체결했다. 삼일제약은 아멜리부 판매로 100억원 상당의 매출을 추가 확보할 것으로 관측된다. 판매 시기는 확정되지 않았다. 지난해말에는 비아트리스 코리아와의 우울제 졸로푸트 등 중추신경계(CNS) 의약품 3종에 대한 국내 유통 판매 계약 맺었다. 해당 품목군 매출은 연간 130억원대로 알려졌다. 성장 동력 방점은 올 9월 완공이 예고된 베트남 점안제 공장이다. 공장이 가동되면 삼일제약의 CMO 사업이 본격화된다. 미국 FDA가 인정하는 cGMP 및 EUGMP급 공장이다. 가동시 연간 1회용 점안제 1.4억관 및 다회용 점안제 0.5억병을 생산할 수 있다. 회사는 추후 글로벌 판매 수요에 따라 공장 유휴부지에 증설도 계획하고 있다. 현재 공장 준공 시점과 맞물려 글로벌 제약사와 점안제 CMO 사업 논의가 이뤄지고 있는 것으로 알려졌다. 시장 관계자는 "삼일제약이 2014년 한국엘러간과 합작법인 청산 후 부침을 겪었지만 최근 성장동력 확보로 반등 조짐을 보이고 있다. 베트남 공장 본격 가동은 호실적에 부스터를 달아줄 것"이라고 평가했다. 베트남 공장 관련 투여 자금은 2018년 유상증자(173억원), 2019년 전환사채(300억원), 교환사채(50억원), 2021년 신주인수권부사채(350억원) 등으로 총 853억원을 유치했다. 기발행 미상환 사채권(473억원 규모) 중 전환사채 잔액(118억원)에 대한 리스크는 최근 교환사채(40억원), 전환사채(50억원) 등을 통해 대비한 상태다. 교환사채는 자사주 일부를 활용했다. 사실상 자사주 매각이다.2022-08-02 06:12:10이석준
호실적 삼일제약, 단숨에 연 매출 2천억 돌파 가시권[데일리팜=이석준 기자] 반기 호실적을 낸 삼일제약이 연매출 2000억원 돌파를 가시권에 뒀다. 기존 최대 매출은 지난해 1340억원이다. 영업이익은 2008년(108억원) 이후 첫 100억원을 넘어설 기세다. 부루펜(OTC), ETC, 안과, CNS(중추신경계) 등 전 사업부가 고른 성장을 거둔 덕분이다. 방점은 올해 9월 완공이 예정된 베트남 점안제 공장이다. 공장 본격 가동시 CMO(위탁생산) 사업 확대로 이어진다. 삼일제약 잠정 공시에 따르면, 회사의 개별 기준 올 반기 매출액과 영업이익은 각각 907억원, 52억원이다. 지난해 상반기와 견줘 37.42%, 173.68% 늘었다. 창립 최대 실적이 점쳐진다. 단순 계산시 연간 매출액은 1814억원, 영업이익 104억원이다. 상승세를 감안하면 외형은 첫 2000억원 돌파도 가능해보인다. 매출 2000억원은 기존 최대인 지난해 1340억원보다 50% 가까이 늘은 수치다. 영업이익은 2008년(108억원) 이후 첫 100억원을 넘기게 된다. 반기 호실적은 '포리부틴', '리박트과립' 등 ETC 주요 품목과 안과, CNS, 위수탁 사업부 등 전 사업부의 고른 성장에 힘입은 결과다. 코로나 여파로 어린이해열제 '어린이부루펜시럽' 판매량도 급증했다. 상반기 매출만 39억원 규모로 전년(28억원)을 넘어섰다. 회사 관계자는 "안정적 재고 확보를 위해 월평균 판매량 이상 재고를 확보하도록 노력하고 있다. 전사 휴가 중에도 어린이부루펜시럽 생산라인의 일부 인원이 근무하여 재고 확보를 위해 총력을 기하고 있다"고 말했다. 잇단 유통 계약…9월 준공 베트남 공장 방점 신규 라인업 보강으로 추가 동력도 마련했다. 회사는 최근 삼성바이오에피스와 황반변성, 당뇨병성 황반부종 치료제 '아멜리부' 국내 독점 유통 판매를 체결했다. 삼일제약은 아멜리부 판매로 100억원 상당의 매출을 추가 확보할 것으로 관측된다. 판매 시기는 확정되지 않았다. 지난해말에는 비아트리스 코리아와의 우울제 졸로푸트 등 중추신경계(CNS) 의약품 3종에 대한 국내 유통 판매 계약 맺었다. 해당 품목군 매출은 연간 130억원대로 알려졌다. 성장 동력 방점은 올 9월 완공이 예고된 베트남 점안제 공장이다. 공장이 가동되면 삼일제약의 CMO 사업이 본격화된다. 미국 FDA가 인정하는 cGMP 및 EUGMP급 공장이다. 가동시 연간 1회용 점안제 1.4억관 및 다회용 점안제 0.5억병을 생산할 수 있다. 회사는 추후 글로벌 판매 수요에 따라 공장 유휴부지에 증설도 계획하고 있다. 현재 공장 준공 시점과 맞물려 글로벌 제약사와 점안제 CMO 사업 논의가 이뤄지고 있는 것으로 알려졌다. 시장 관계자는 "삼일제약이 2014년 한국엘러간과 합작법인 청산 후 부침을 겪었지만 최근 성장동력 확보로 반등 조짐을 보이고 있다. 베트남 공장 본격 가동은 호실적에 부스터를 달아줄 것"이라고 평가했다. 베트남 공장 관련 투여 자금은 2018년 유상증자(173억원), 2019년 전환사채(300억원), 교환사채(50억원), 2021년 신주인수권부사채(350억원) 등으로 총 853억원을 유치했다. 기발행 미상환 사채권(473억원 규모) 중 전환사채 잔액(118억원)에 대한 리스크는 최근 교환사채(40억원), 전환사채(50억원) 등을 통해 대비한 상태다. 교환사채는 자사주 일부를 활용했다. 사실상 자사주 매각이다.2022-08-02 06:12:10이석준 -
 녹십자, 2Q 매출 9%↑...백신·혈액제제·처방약 호조[데일리팜=천승현 기자] 녹십자가 백신, 혈액제제, 처방의약품 등 전 사업부가 성장하며 실적 호조를 나타냈다. 녹십자는 지난 2분기 영업이익이 131억원으로 전년동기대비 18.0% 늘었다고 1일 공시했다. 매출액은 4232억원으로 전년보다 9.2% 증가했고 당기순이익은 109억원으로 45.3% 늘었다. 2분기 매출은 작년 3분기의 4657억원에 이어 역대 두 번째 규모다. 2분기 매출만 보면 창립 이후 최대 규모다. 혈액제제 매출이 15% 증가한 1060억원을 기록했고 백신 매출은 844억원으로 전년대비 8% 확대됐다. 회사 측은 "역대 최대치를 기록한 남반구향 독감백신은 664억원의 매출을 올렸으며 혈액제제 해외 매출도 판매량 확대 및 단가 인상으로 두자릿 수 성장세를 나타냈다"라고 설명했다. 처방의약품 매출은 811억원으로 전년대비 5% 늘었고 소비자헬스케어 등 기타 부문이 지난해보다 8% 성장한 509억원을 나타냈다. 미국에서 개발 중인 차세대 대상포진 백신 임상 2상의 영향으로 연구개발비가 전년 대비 대폭 증가했지만 주력 제품 호실적을 통해 수익성은 개선됐다. 녹십자는 “3분기부터는 북반구 독감백신 매출이 인식되면서 하반기에도 백신 부문 매출 호조세가 지속될 것”이라고 내다봤다. 앞서 실적을 발표한 연결 대상 계열사들도 준수한 성장세를 기록했다. GC셀은 검체검사와 바이오물류 사업이 지속 성장하며 2분기 매출 557억원, 영업이익 50억원으로 견조한 실적을 냈다. GC녹십자웰빙도 주사제 및 건기식 사업 호조로 두자릿 수의 매출 외형 성장을 기록했다. 반면 GC녹십자엠에스는 진단 키트 사업 부진의 영향으로 역성장했다.2022-08-01 15:04:01천승현
녹십자, 2Q 매출 9%↑...백신·혈액제제·처방약 호조[데일리팜=천승현 기자] 녹십자가 백신, 혈액제제, 처방의약품 등 전 사업부가 성장하며 실적 호조를 나타냈다. 녹십자는 지난 2분기 영업이익이 131억원으로 전년동기대비 18.0% 늘었다고 1일 공시했다. 매출액은 4232억원으로 전년보다 9.2% 증가했고 당기순이익은 109억원으로 45.3% 늘었다. 2분기 매출은 작년 3분기의 4657억원에 이어 역대 두 번째 규모다. 2분기 매출만 보면 창립 이후 최대 규모다. 혈액제제 매출이 15% 증가한 1060억원을 기록했고 백신 매출은 844억원으로 전년대비 8% 확대됐다. 회사 측은 "역대 최대치를 기록한 남반구향 독감백신은 664억원의 매출을 올렸으며 혈액제제 해외 매출도 판매량 확대 및 단가 인상으로 두자릿 수 성장세를 나타냈다"라고 설명했다. 처방의약품 매출은 811억원으로 전년대비 5% 늘었고 소비자헬스케어 등 기타 부문이 지난해보다 8% 성장한 509억원을 나타냈다. 미국에서 개발 중인 차세대 대상포진 백신 임상 2상의 영향으로 연구개발비가 전년 대비 대폭 증가했지만 주력 제품 호실적을 통해 수익성은 개선됐다. 녹십자는 “3분기부터는 북반구 독감백신 매출이 인식되면서 하반기에도 백신 부문 매출 호조세가 지속될 것”이라고 내다봤다. 앞서 실적을 발표한 연결 대상 계열사들도 준수한 성장세를 기록했다. GC셀은 검체검사와 바이오물류 사업이 지속 성장하며 2분기 매출 557억원, 영업이익 50억원으로 견조한 실적을 냈다. GC녹십자웰빙도 주사제 및 건기식 사업 호조로 두자릿 수의 매출 외형 성장을 기록했다. 반면 GC녹십자엠에스는 진단 키트 사업 부진의 영향으로 역성장했다.2022-08-01 15:04:01천승현 -
녹십자, 2Q 영업익 131억...전년비 18%↑[데일리팜=천승현 기자] 녹십자는 지난 2분기 영업이익이 131억원으로 전년동기대비 18.0% 늘었다고 1일 공시했다. 매출액은 4232억원으로 전년보다 9.2% 증가했고 당기순이익은 109억원으로 45.3% 늘었다.2022-08-01 14:32:59천승현
-
 에스티팜, 올리고 제조소 美 FDA cGMP 인증 획득[데일리팜=정새임 기자] 에스티팜은 아시아 최초로 미국 식품의약국(FDA)으로부터 올리고 제조소 우수의약품품질관리기준(cGMP) 인증을 지난달 29일에 획득했다고 1일 밝혔다. FDA 실사단은 지난 5월 16일부터 20일까지 5일간 에스티팜 반월 캠퍼스 올리고동을 방문해 '신약 승인 전 제조소 실사(Pre Approval Inspection, PAI)를 진행했다. PAI 실사는 신약 허가 및 원료의약품 공급을 위해 반드시 필요한 절차다. 에스티팜은 실사 결과 문제점이 발견되지 않고 보정 자료 제출이 필요 없는 무결점(No Action Indicated, NAI) 등급을 받았다. 에스티팜은 지금까지 미국 시장에 임상용 올리고핵산치료제 원료의약품 수출만 가능했다. 이번 FDA cGMP 승인으로 회사는 미국 시장에 대규모 상업화 물량 수출이 가능해진다. 에스티팜은 2023년 상반기까지 4건의 FDA PAI실사가 예정되어 있다. 회사는 원료를 공급하는 신약들의 FDA 승인이 임박했음을 의미한다고 설명했다. 에스티팜 관계자는 "아시아 최초로 올리고 생산설비에 대한 FDA PAI실사를 통과했고 실사 결과 문제점이 발견되지 않은 무결점 등급의 인증을 받았다는 점에서, 에스티팜의 cGMP 역량을 다시 한번 입증하게 됐다"며 "이번 FDA cGMP 승인이 향후 진행될 실사에도 긍정적인 영향을 미칠 것으로 예상되며, 해당 신약들이 상업화에 성공하면 에스티팜의 매출이 큰 폭으로 성장할 것으로 전망된다"고 말했다.2022-08-01 14:12:41정새임
에스티팜, 올리고 제조소 美 FDA cGMP 인증 획득[데일리팜=정새임 기자] 에스티팜은 아시아 최초로 미국 식품의약국(FDA)으로부터 올리고 제조소 우수의약품품질관리기준(cGMP) 인증을 지난달 29일에 획득했다고 1일 밝혔다. FDA 실사단은 지난 5월 16일부터 20일까지 5일간 에스티팜 반월 캠퍼스 올리고동을 방문해 '신약 승인 전 제조소 실사(Pre Approval Inspection, PAI)를 진행했다. PAI 실사는 신약 허가 및 원료의약품 공급을 위해 반드시 필요한 절차다. 에스티팜은 실사 결과 문제점이 발견되지 않고 보정 자료 제출이 필요 없는 무결점(No Action Indicated, NAI) 등급을 받았다. 에스티팜은 지금까지 미국 시장에 임상용 올리고핵산치료제 원료의약품 수출만 가능했다. 이번 FDA cGMP 승인으로 회사는 미국 시장에 대규모 상업화 물량 수출이 가능해진다. 에스티팜은 2023년 상반기까지 4건의 FDA PAI실사가 예정되어 있다. 회사는 원료를 공급하는 신약들의 FDA 승인이 임박했음을 의미한다고 설명했다. 에스티팜 관계자는 "아시아 최초로 올리고 생산설비에 대한 FDA PAI실사를 통과했고 실사 결과 문제점이 발견되지 않은 무결점 등급의 인증을 받았다는 점에서, 에스티팜의 cGMP 역량을 다시 한번 입증하게 됐다"며 "이번 FDA cGMP 승인이 향후 진행될 실사에도 긍정적인 영향을 미칠 것으로 예상되며, 해당 신약들이 상업화에 성공하면 에스티팜의 매출이 큰 폭으로 성장할 것으로 전망된다"고 말했다.2022-08-01 14:12:41정새임
-
 [SK바이오팜] CNS Research Center 연구원 경력 모집
[SK바이오팜] CNS Research Center 연구원 경력 모집 -
 해외사업본부 PIS팀 약사 채용(경력무관)
해외사업본부 PIS팀 약사 채용(경력무관) -
 국립교통재활병원 정규직 약사 채용공고
국립교통재활병원 정규직 약사 채용공고 -
 계명대학교 동산의료원 2026학년도 임시직(야간약사) 계약직원 상시 공개채용 공고
계명대학교 동산의료원 2026학년도 임시직(야간약사) 계약직원 상시 공개채용 공고 -
 [더자인병원] 원내 상근 약사 초빙합니다.
[더자인병원] 원내 상근 약사 초빙합니다. -
 연세대학교 의료원 신규 약사(정규직) 모집
연세대학교 의료원 신규 약사(정규직) 모집 -
 [강동성심병원] 약사 채용공고(ASP전담 / 주말 약사)
[강동성심병원] 약사 채용공고(ASP전담 / 주말 약사) -
 경상북도 김천의료원 약사 채용
경상북도 김천의료원 약사 채용 -
 약제팀 정규약사(신규/경력) 모집
약제팀 정규약사(신규/경력) 모집 -
 동아대학교병원 약제부 약사 <정규직> 모집
동아대학교병원 약제부 약사 <정규직> 모집 -
 [광주보훈병원] 정규직 약무직(약사) 공개채용
[광주보훈병원] 정규직 약무직(약사) 공개채용 -
 제조관리 약사 채용 신입/경력
제조관리 약사 채용 신입/경력 -
 야간 계약직(금/일) / 주간 정규직·계약직 약사 공채
야간 계약직(금/일) / 주간 정규직·계약직 약사 공채 -
 [서울 30분] 한림병원 약제센터 야간 약사모집
[서울 30분] 한림병원 약제센터 야간 약사모집 -
 평택공장 제조관리약사 채용(경력무관)
평택공장 제조관리약사 채용(경력무관) -
 세종 공장 품질관리약사 (3년↑)
세종 공장 품질관리약사 (3년↑) -
 개발팀 PV/MA/제제연구 부문별 경력채용
개발팀 PV/MA/제제연구 부문별 경력채용 -
 광주광역시 동구에서 원내약국 약사님을 모십니다~
광주광역시 동구에서 원내약국 약사님을 모십니다~ -
 일산차병원 약제팀 주간약사 채용
일산차병원 약제팀 주간약사 채용 -
 [일산백병원] 약제부 계약직 야간/주말당직약사 (신입/경력) 모집 공고
[일산백병원] 약제부 계약직 야간/주말당직약사 (신입/경력) 모집 공고 -
 환인제약(주) 6월 신입/경력 채용 (~6/14(일))
환인제약(주) 6월 신입/경력 채용 (~6/14(일)) -
 명인제약 【상반기】 경력사원 모집
명인제약 【상반기】 경력사원 모집 -
 당진공장 품질관리약사 채용(✨신입우대)
당진공장 품질관리약사 채용(✨신입우대) -
 가천대길병원 약제부 야간약사(계약직) 채용
가천대길병원 약제부 야간약사(계약직) 채용 -
 전북특별자치도마음사랑병원 주간약사 채용공고
전북특별자치도마음사랑병원 주간약사 채용공고 -
 의정부/노원을지대학교병원 약사 채용
의정부/노원을지대학교병원 약사 채용



오늘의 TOP 10
- 1도수치료 연 최대 24회 제한…회당 4만원대 관리급여 적용
- 2유나이티드제약 '클란자CR정' 러시아 무기한 품목 허가
- 3퍼스트바이오, 빅파마 출신 SAB 꾸려 신약개발 속도
- 4종근당 천연물 위염치료제 '지텍' 조건부 약평위 통과
- 5공정위, ‘탁소텔 인수’ 보령에 제네릭 매각 시정조치
- 6동광 "회수대상 인데놀은 허가변경 전 제품…불순물 관련 없어"
- 7파마리서치, 해양 정화 활동 전개…ESG 실천 강화
- 8CJ웰케어, IHMC서 균주 맞춤형 포뮬러 기술 공개
- 9데일리팜 이정환·정흥준 기자, 인신협 이달의 기자상 우수상
- 10에이비엘바이오, 이중항체 면역항암제 후보 병용 임상 확대