














생물의약품 GMP 지침 신설…합성약과 분리 운영
- 이정환
- 2016-07-05 06:14:51
-
가
- 가
- 가
- 가
- 가
- 가
-




- 식약처 "적합판정 제조소 실사면제 3년으로 확대"
- PR
- 전국 지역별 의원·약국 매출&상권&입지를 무료로 검색하세요!!
- 데일리팜맵 바로가기

지금까지 부재했던 바이오약 GMP 사전검토 지침은 새로 만들었다. 합성약과 바이오약으로 분류·재편중인 제약산업 패러다임에 발 맞춰 품질관리기준을 선진화하기 위한 것이다.
4일 식약처 관계자는 "바이오약 허가신청 GMP 평가지침을 별도 마련하고, 사건검토 지침도 신설했다"고 설명했다.
분할되면서 변경된 중요사안은 식약처로부터 바이오약 GMP 평가 결과 적합 판정된 원료·완제약 제조소 내 작업소(바이오약 생산공장)는 향후 3년까지 서류제출로 현장실사를 갈음할 수 있게 된 점이다. 사실상 현지실사를 3년 간 면제받게 된 것.
기존에는 적합 판정받은 작업소라도 업체별 1~2년 내 식약처 현장실사를 받아야 했다. 물론 평가자료 제출이 어려운 경우 실태조사를 신청하면 식약처 GMP 조사관 2~5명이 생산공장을 방문해 실사를 진행한다.
그러나 이번 조치로 GMP 적합 제약사들은 기존보다 완화된 실사 기준을 적용받을 수 있게 됐다. 실질적으로는 실사비용이 줄고 불필요한 행정력 낭비를 줄일 수 있게된 셈이다.
아울러 품목 변경이나 허가 목적 바이오약의 GMP사전검토 가이드라인도 새로 생겼다. 제조소 평면도 등 11종 제출서류 평가를 기본으로 필요한 경우 실태조사가 더해진다.
실태조사 대상은 최초 평가대상 작업소에서 만들어지는 바이오의약품이다.
제약사가 바이오약 GMP 사전검토를 신청하면, 식약처와 식품의약품안전평가원 소속 사무관·주무관(심사관), 관할 지방식약청 소속 조사관으로 구성된 '사전검토 팀'이 마련돼 이번에 분리된 '바이오약 허가신청 GMP 평가지침'에 의거해 실사가 이뤄진다.
해외 소재 제조원 실태조사가 필요한 경우 해외실사 비용은 수익자인 신청 제약사가 부담해야 한다.
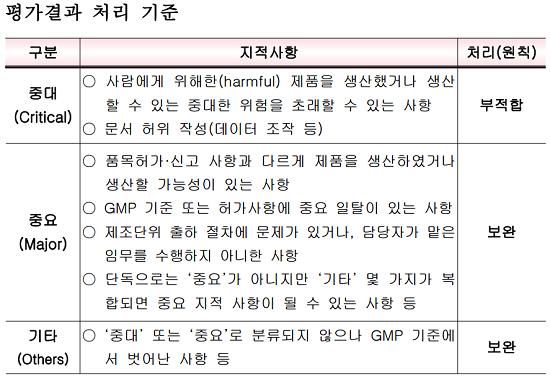
이 밖에 바이오약 GMP 평가기준은 과거와 동일하다. 평가결과 사람에게 위해한 제품을 생산했거나 데이터 조작 등 문서를 허위 작성하는 중대(critical) 지적사항이 발생하면 부적합 판정을 받는다.
허가·신고 사항과 다른 제품을 생산했거나 주요 GMP 사항에 일탈이 발생하는 중요(major) 지적사항의 경우 보완 처리된다. 중대하지 않은 GMP 기준을 위반해도 기타(others) 지적사항에 따라 동일한 조치가 내려진다.
식약처 관계자는 "바이오약 비중이 높아짐에 따라 합성약과 구분해 GMP 품질관리기준 가이드라인 분리 필요성이 생겼다"며 "적합 판정 작업소는 3년간 서류로 현지실사를 갈음하도록 규제를 합리화한 게 핵심"이라고 설명했다.
- 익명 댓글
- 실명 댓글
- 댓글 0
- 최신순
- 찬성순
- 반대순
-
 정규직 2026년 신입 약사 채용
정규직 2026년 신입 약사 채용 -
 개발팀 PV/MA/제제연구 부문별 경력채용
개발팀 PV/MA/제제연구 부문별 경력채용 -
 계명대학교 동산의료원 2026학년도 임시직(야간약사) 계약직원 상시 공개채용 공고
계명대학교 동산의료원 2026학년도 임시직(야간약사) 계약직원 상시 공개채용 공고 -
 한국에자이 QA Associate 채용
한국에자이 QA Associate 채용 -
 [더자인병원] 원내 상근 약사 초빙합니다.
[더자인병원] 원내 상근 약사 초빙합니다. -
 Senior Manager, Government Affairs & External Liaison
Senior Manager, Government Affairs & External Liaison -
 삼성서울병원 약제부 야간전담 약사 채용
삼성서울병원 약제부 야간전담 약사 채용 -
 [부산보훈병원] '26년 정규직 약사 공개채용
[부산보훈병원] '26년 정규직 약사 공개채용 -
 전북특별자치도마음사랑병원 주간약사 채용공고
전북특별자치도마음사랑병원 주간약사 채용공고 -
 울진군의료원 약사 채용 공고
울진군의료원 약사 채용 공고 -
 관리약사/주사제 제조/영업 담당자 모집
관리약사/주사제 제조/영업 담당자 모집 -
 한국의약품안전관리원 개방형 직위 (의약품안전조사본부장) 채용 재공고
한국의약품안전관리원 개방형 직위 (의약품안전조사본부장) 채용 재공고 -
 [구미차병원] 약제팀 야간약사 모집
[구미차병원] 약제팀 야간약사 모집 -
 [강동성심병원] 약사 채용공고(ASP전담 / 정규직 약사)
[강동성심병원] 약사 채용공고(ASP전담 / 정규직 약사) -
 [오노파마코리아] RA Specialist 채용(계약직)
[오노파마코리아] RA Specialist 채용(계약직) -
 [서울 30분] 약제센터 야간 약사모집
[서울 30분] 약제센터 야간 약사모집 -
 CV 마케팅부 PM 경력사원 채용
CV 마케팅부 PM 경력사원 채용 -
 로뎀요양병원 약사님 모십니다.
로뎀요양병원 약사님 모십니다. -
 [일산백병원] 약제부 계약직 야간약사 (신입/경력) 모집 공고
[일산백병원] 약제부 계약직 야간약사 (신입/경력) 모집 공고 -
 서울대학교병원 임상시험센터 약국 연구원 약사 채용 공고
서울대학교병원 임상시험센터 약국 연구원 약사 채용 공고 -
CV부 MR 정규직 경력사원 채용(창원 지역)
-
 경남 함안공장 제조관리약사 책임자 채용
경남 함안공장 제조관리약사 책임자 채용 -
 삼성서울병원 (주말전담) 약제부 계약직 약사 채용
삼성서울병원 (주말전담) 약제부 계약직 약사 채용 -
 해외사업본부 PIS팀 약사 채용
해외사업본부 PIS팀 약사 채용 -
 당진공장 품질관리약사 채용(경력무관)
당진공장 품질관리약사 채용(경력무관) -
 5/29(금) 하루 근무하실 약사님 연락부탁드립니다~
5/29(금) 하루 근무하실 약사님 연락부탁드립니다~ -
Specialist, Hematology and Oncology Marketing
-
 의정부/노원을지대학교병원 약사 채용
의정부/노원을지대학교병원 약사 채용 -
 약제본부 야간약사 채용 안내
약제본부 야간약사 채용 안내 -
 일산차병원 약제팀 주간약사 채용
일산차병원 약제팀 주간약사 채용 -
 [바이엘코리아] MSL I Radiology (약 18개월 육아휴직 대체 계약직, 공고 연장)
[바이엘코리아] MSL I Radiology (약 18개월 육아휴직 대체 계약직, 공고 연장)

약국e몰
![[종근당] 브레이닝캡슐](https://cdn.platpharm.co.kr/2025/06/2506040708450012544.png)
![[일양약품] 도담도담 시리즈](https://cdn.platpharm.co.kr/2024/02/2402020935180000240.jpg)
![[리쥬올] PDLLA 퍼밍 크림 30ml](https://cdn.platpharm.co.kr/2026/04/2604070229110000386.webp)
![[경방신약] 방콜브이산](https://cdn.platpharm.co.kr/2025/12/2512310630020002495.webp)
![[신신제약] 아렉스마일드](https://cdn.platpharm.co.kr/2023/11/2311300927130000133.jpg)
![[유한양행] 안티푸라민 파스 시리즈](https://cdn.platpharm.co.kr/2024/05/2405280631070000069.png)
![[신신제약] 모스키토 밀크](https://cdn.platpharm.co.kr/2025/10/2510150733400004067.webp)
![[삼진제약] 게보핏 시리즈](https://cdn.platpharm.co.kr/2024/07/2407100728250000386.png)
![[유한양행] 마그비 시리즈](https://cdn.platpharm.co.kr/2024/03/2403261023280000193.jpg)
![[노보노디스크] 위고비](https://cdn.platpharm.co.kr/static/dailypharm/Wigobi.png)
![[일양약품] 프로엑스피](https://cdn.platpharm.co.kr/2026/01/2601221008450010125.webp)
![[옵투스] 오에수 시리즈](https://cdn.platpharm.co.kr/2026/02/2602130209000031633.webp)
![[아워팜] 우리아이 맞춤설계, 바로타민 kids 엘더베리맛](https://i.baropharm.com/partner/products/3f39593e-6318-4dd9-a778-c008c868b5c8.png)
![[한독] 붙이는 통증 전문가, 케토톱 액티브 플라스타(쿨) 40매](https://i.baropharm.com/products/202503/1741829602305.png)
![[알엑스미] 알엑스미 리쥬영 울트라 PDRN 10000 딥리페어 크림](https://i.baropharm.com/partner/products/70c72dd0-cfd3-4d80-87e4-dc4f8de6658b.png?label=바뷰페로고)
![[레비온] PDRN+EGF, 레비온RX PDRN EGF 크림](https://i.baropharm.com/products/202512/1765949426601.png)
![[켄뷰] 오리지널 폼타입, 로게인5%폼에어로졸60g](https://i.baropharm.com/products/dc84d96e-d0b4-46bc-bcc8-d62016406fe4.png)
![[경동제약] 인태반 자양강장제, 파워콤프](https://i.baropharm.com/partner/products/0a2cbb4c-96c5-40a5-aec2-8beeae11682c.png)
![[휴온스 ] 비듬을 한번에, 니조랄 2%액](https://i.baropharm.com/products/478a284d-4361-4b4a-8a00-8bab80f34319.png?label=PLAN_01)
![[켄뷰] 다양한 통증에, 타이레놀정 500mg 10정](https://i.baropharm.com/products/6c6ea4f4-7ab2-44f2-a165-f062d80f525b.png)
![[CHD제약] 50년 전통의 천혜당 식염포도당](https://i.baropharm.com/products/202604/1775095858899.png)
![[쥬베룩] 진짜 쥬베룩을 담은 약국전용 PDLLA 크림](https://i.baropharm.com/products/202604/1775343960671.png?label=바뷰페로고)
![[아워팜] CJ웰케어, 바이오코어 1000억 유산균](https://i.baropharm.com/products/202604/1776750298620.png)
![[리쥬올] 닥터 리쥬올 어드밴스드 PDRN 리쥬비네이팅 크림 30ml](https://i.baropharm.com/partner/products/a201d2b4-f21e-4b13-957c-846d286b3d21.jpg?label=바뷰페로고)
오늘의 TOP 10
- 1CSO 규제 향방은…복지부, 재위탁·수수료율 손질 가능성
- 2시골 청년서 900억 기업 일군 파마피아 문규연대표의 뚝심
- 3공정위, 가격통제 제재…약국 전용 건기식 유통 지각변동?
- 4하나제약, 삼진제약 5년 투자 헛심…원금 수준 투자금 회수
- 5부광, 4년째 공장가동률 100%↑…시급한 유니온 인수 타이밍
- 6네트워크 약국 방지법 오늘 공포…11월 27일부터 시행
- 7중동전쟁 영향 미쳤나…제약, 수액제 원부자재 매입 감소
- 8아미반타맙+레이저티닙, 수술 전 선행보조요법까지 확장
- 9[기자의눈] 약가유연계약, 실제가 제공 범위 고민해야
- 10유방암 표적 치료 'CDK4/6억제제' 급여 확대 시험대




