- LOGIN
- MemberShip
- 2026-05-04 10:36:30
- Policy
- Roche Korea speeds up reimb of Ocrevus in KOR
- by Lee, Tak-Sun Jul 12, 2024 05:48am
- Roche Korea is making active moves to receive reimbursement for its multiple sclerosis drug Ocrevus, which was approved in May this year. Roche, which applied for reimbursement immediately after the approval, has set out to persuade the Health Insurance Review and Assessment Service to expedite the application process. According to industry sources, Roche Korea recently applied for a drug presentation session to HIRA. The system was introduced in 2010 to increase the transparency and objectivity of drug evaluations by sharing information about new drugs between pharmaceutical companies and reviewers. The briefing is held between 1-2 months after the company applies for evaluations. However, for drugs that require supplemental data, a briefing is held after the supplemental data has been submitted and the reviewers have understood the submitted data. The briefing is open to reviewers and deputy directors involved in the evaluation of the reimbursement standard and the evaluation of the new drug. Roche requested to hold the briefing to provide reviewers with an accurate representation of the drug's efficacy and clinical utility and justify the drug’s reimbursement. Ocrevus is considered to have dramatically improved dosing convenience for patients with multiple sclerosis. As a recombinant humanized monoclonal antibody (mAb, IgG1) that selectively targets CD20-expressing B cells, it reduces the number and function of B cells, thereby inhibiting MS. MS is a chronic disease in which myelin sheaths are damaged by an autoimmune inflammatory response. Damage to the myelin sheath causes symptoms such as muscle weakness, fatigue, and vision impairment, and can lead to non-traumatic neurological disability. An estimated 1,800 patients are known to be living with MS in Korea. Previously, patients had to receive weekly injections such as beta interferon to relieve their symptoms. Ocrevus, on the other hand, is said to be more convenient to administer as the first dose of 600 mg is divided into 2 intravenous infusions, and the 600 mg doses thereafter are administered as a single intravenous infusion every 6 months. The drug was approved by the U.S. FDA in 2017 and its subcutaneous formulation recently received EU approval. A barrier to its reimbursement in Korea is the high price of the drug. In the U.S., the annual cost of the drug is nearly KRW 100 million. Although it is expensive, the company likely applied for the briefing to emphasize the justification for its reimbursement. In particular, there are observations that Roche may be rushing to receive reimbursement as Celltrion is avidly developing a biosimilar of the drug. An industry insider explained, "Through the drug briefing, pharmaceutical companies can appeal to not only the reviewers but also the relevant deputy directors on how just the reimbursement is, and share complementary ideas on key issues, which can speed up the submission of documents in the future reimbursement review process."
- Company
- Protein C deficiency drug 'Ceprotin' begins negotiations
- by Eo, Yun-Ho Jul 12, 2024 05:48am
- Takeda Pharmaceuticals Korea 'Ceprotin' has entered the last stage of obtaininig reimbursement listing. According to our investigation, Takeda Pharmaceuticals Korea has started to negotiate drug pricing with the National Health Insurance Service (NHIS) for Ceprotin (Human potein C), a novel drug for the treatment of congenital protein C deficiency. ‘Human protein C deficiency’ is a type of thrombotic disease that causes blood clotting and it was first reported in 1981. 'Severe congenital protein C deficiency,' a type of protein C deficiency, is an extremely rare disease that occurs in one in every 4 million. In South Korea, there have been reported cases of 13 patients to date (according to 2019 KOSIS statistics). Congenital protein C deficiency is a hereditary disorder that occurs when the body lacks ‘protein c,’ a naturally occurring thrombolytic agent in the body, due to genetic abnormalities. On average, healthy individuals have a 'protein C' level of around 100 ng/dL. However, patients with severe congenital protein C deficiency are known to have less than 1% of normal levels (less than 1 ng/dL), disrupting the balance between blood clotting and anticoagulation. This imbalance results in frequent thrombosis, recurrent venous thrombosis, venous stasis ulcers, and pulmonary embolism. These symptoms can lead to high-risk conditions such as myocardial infarction or stroke. Additionally, it occurs when blood vessels become blocked due to thrombosis, causing bleeding throughout the body, the most common manifestation is large, purplish patches resembling bruises on the skin. Currently, there is no known drug therapy that can prevent or fully control venous thrombosis and purpura fulminans (PF) in patients with severe congenital protein C deficiency. Ceprotin is a concentrated solution of human protein C made from human protein C. It received the Orphan Drug Designation from the Ministry of Food and Drug Safety (MFDS) in March 2021. Based on an early phase clinical study, enrolling 18 patients with severe congenital protein C deficiency, Ceprotin was demonstrated to be effective in treating purpura fulminans (PF) or other thrombotic events compared to the histological control group. In the study, 18 patients with purpura fulminans (PF) (6 severe, 11 moderate, and 1 mild) were treated with Ceprotin. 17 patients (94.4%) were assessed as 'effective,' 1 patient (5.6%) as 'effective with complications.' There were no reports of 'ineffective' outcomes. This result demonstrated more effective treatment outcomes than a control group (21 patients), which received traditional therapies such as fresh frozen plasma or traditional anticoagulants. In the secondary efficacy rating of Ceprotin, out of 18 cases of purpura fulminans (PF), 13 cases (72.2%) were evaluated as 'excellent,' 4 cases (22.2%) as 'good,' and 1 case (5.6%) of severe purpura fulminans (PF) was assessed as 'fair.' Additionally, out of the 5 cases of venous thrombosis, 4 cases (80%) were evaluated as 'excellent' and 1 case (20%) as 'good'. Furthermore, Ceprotin effectively reduced the size and number of skin lesions. Non-necrotic skin lesions were treated within a maximum of 12 days (median 4 days), while necrotic skin lesions were treated within a maximum of 52 days (median 11 days) with this drug.

- Policy
- Reassessing the plan for comparing foreign drug prices
- by Lee, Tak-Sun Jul 12, 2024 05:48am
- The government will likely reassess foreign drug price comparison by evaluating the therapeutic category with the highest number of products. Consequently, gastrointestinal agents, high blood pressure drugs, and antibiotics will be assessed in the first year. There are over 2000 products in these three therapeutic categories. Furthermore, drugs with the same ingredient products produced by fewer than three companies will likely to be excluded from the assessment. According to industry sources on July 10th, the government is gathering opinions from pharmaceutical companies about the current reassessment plan, which has been shared through ten meetings with the pharmaceutical industry (as of July 5th). The current reassessment plan is not the final one. The issue is still ongoing because it does not include the referencing sources of Germany and Canada. In the first year, according to the reassessment plan, therapeutic categories with the highest number of products, including gastrointestinal agents (2043 products), high blood pressure drugs (2268 products), and antibiotics (2156 products), will be assessed. In the second year, hyperlipidemia drugs, respiratory system medications, central nervous system medications, diabetes drugs, and musculoskeletal disorder medications will be assessed. In the third year, ophthalmology drugs, otolaryngology drugs, dentistry drugs, painkillers, urological and reproductive system medications, anticoagulants, dermatological treatments, anticancer agents, and 17 other therapeutic categories will be assessed. The current plan to reassess foreign drug price comparison (as of July 5th). (Therapeutic category with the highest number of products) In the first year, gastrointestinal agents (2043 products), high blood pressure drugs (2268 products), and antibiotics (2156 products), will be assessed. In the second year, hyperlipidemia drugs, respiratory system medications, central nervous system medications, diabetes drugs, and musculoskeletal disorder medications will be assessed. In the third year, ophthalmology drugs, otolaryngology drugs, dentistry drugs, painkillers, urological and reproductive system medications, anticoagulants, dermatological treatments, anticancer agents, and 17 other therapeutic categories will be assessed. However, exclusion will be applied to ▲low-priced drugs, orphan drugs, drugs that have been listed as shortage prevention drugs (SPD) ▲Oxygen, nitrogen dioxide, saline solutions, artificial perfusion agents, and radioactive drugs ▲Narcotics ▲Drugs with the same administration, ingredients, and formulation produced by fewer than three companies ▲Products undergone price increases (after January 2020). During the discussion, drugs with the same administration, ingredients, and formulation produced by fewer than three companies were added to the list. A reference price for adjustment is the average price of drugs found in A8 countries. Adjusted mean price will exclude the highest and the lowest prices. Products with a higher drug price than the calculated reference amount will be subjected to a reduction. In cases where fewer than 2 out of 8 countries can be found, the average reduction rate of the most similar product is applied, considering factors such as content, ingredients, formulations, and administration methods. Products with generic prices higher than the standard amount calculated using the highest price within the same product will be reduced. When calculating the average reduction rate, if a negative reduction rate occurs due to lower prices compared to foreign countries, it will be reflected as such in the calculation. For combination therapies, if the assessed amount is lower than the sum of the assessed amounts for monotherapy or combination therapy, the reduction will be limited to the sum of the amounts assessed. The prices of pharmaceuticals that have submitted the required documents will be reduced to the amount assessed for their intended development products. Furthermore, it has been revealed that the current plan included the addition of criteria stating that the calculation of price adjustments referencing foreign drug prices should be based on public prices reimbursed or reimbursed equivalently in the respective country. The pharmaceutical industry's significant opposition to the public reimbursement requirements during the tenth meeting may prompt revisions in the final plan.
- Company
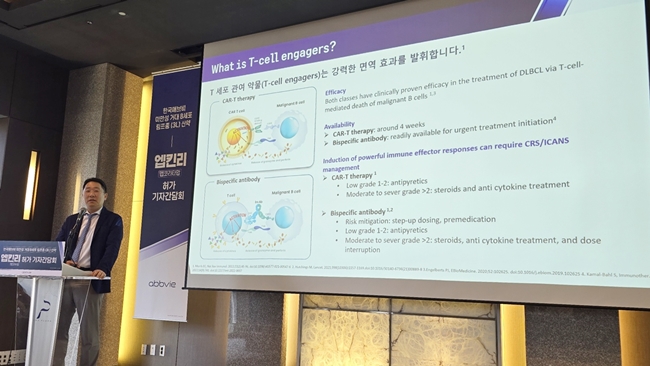
- Will Epkinly become a game-changer in the blood cancer mkt?
- by Hwang, Byung-woo Jul 12, 2024 05:47am
- The arrival of bispecific antibody-based therapies in Korea is expected to shift the blood cancer treatment market paradigm. The industry is welcoming the introduction of a new treatment option, as an unmet need exists in diffuse large B-cell lymphoma (DLBCL), which has a poor prognosis even after three or later lines of treatments. AbbVie hosted a press conference on the 10th to highlight the significance of Epkinly’s approval. Professor Jin Seok Kim. Department of Hematology-Oncology at Severance Hospital, is presenting data related to blood cancer.AbbVie hosted a press conference on the 10th to celebrate the launch of 'Epkinly (epcoritamab)’ as a third-line treatment for DLBCL and highlight the implications of the approval. Epkinly was approved by the Ministry of Food and Drug Safety in late June for the treatment of adult patients with relapsed or refractory DLBCL who have received 2 or more prior systemic therapies. Epkinly is a humanized bispecific antibody (IgG1) that binds to CD20 on B cells and CD3’s extracellular specific epitopes on T cells. It has a mechanism of action that induces specific T-cell activation and T-cell-mediated CD20-expressing cell death by simultaneously acting on CD20-expressing cancer cells and CD3-expressing endogenous T cells. Epkinly is the first subcutaneous bispecific antibody approved in Korea for the treatment of DLBCL. It has the advantage of being administered in less than one minute, allowing for a relatively short hospital stay and rapid treatment. The duration of the treatment is not limited and can be administered until disease progression or unacceptable toxicity Epkinly’s approval is noteworthy because it provides a new option in the treatment of DLBCL, an area with much unmet need. Professor Deok Hwan Yang, Department of Hematology-Oncology at Chonnam National University Hwasun Hospital, said, "Even after receiving first-line standard therapy for DLBCL, 30 to 40% of patients relapse or become refractory to treatment and move on to the next line of treatment. Patients who relapse after receiving autologous stem cell transplantation as a second-line treatment have poor prognosis, and patients who relapse after receiving CAR-T as a third-line treatment also have poor outcomes." This means that patients who relapse after receiving three or more lines of treatment generally have a poor prognosis, with lower overall response rates and worse survival outcomes. "Currently, there are limited options in the third and later lines of treatment, with no set standard of care. In this way, there is a large unmet need for new options.” (From the left) Professor Deok Hwan Yang, Department of Hematology-Oncology at Chonnam National University Hwasun Hospital; Professor Jin Seok Kim. Department of Hematology-Oncology at Severance HospitalThe study that became the basis of Epkinly’s approval is the EPCORE NHL-1 study, a non-randomized, single-arm trial. Its efficacy analysis showed an overall response rate (ORR) of 62%, with 39% achieving complete response (CR). Professor Jin Seok Kim. Department of Hematology-Oncology at Severance Hospital, said, “We found Epkinly was well tolerated in heavily pretreated third-line patients, and most adverse events were manageable and predictable. At 30 months of follow-up, median overall survival (mOS) was 19.4 months, confirming that it is a viable option for prolonging survival in this patient population." The advent of such bispecific antibody-based therapies sheds light on the question of how they will compare to CAR-T therapies that are being reimbursed through national health insurance. Experts predict that bispecific antibody therapies such as Epkinly will become complementary options in the future, assuming that they also be reimbursed. Professor Kim explained, "If you look at the average cost, I think they will be very similar to CAR-T therapies, and I don't think one can be a substitute for the other. I think it's going to be a patient-specific choice because they have different targets, but I think there will be cost concerns."
- Policy
- ‘Bioequivalence tests rather intensified price cuts'
- by Lee, Tak-Sun Jul 12, 2024 05:47am
- The pharmaceutical industry is calling for improvements as their drugs that have completed bioequivalence tests per the government’s insurance price ceiling reevaluations may face larger price cuts under ‘Type C’ of the Price Volume Agreement negotiations this year. Generic drugs that have completed bioequivalence tests during the insurance price ceiling reevaluations have maintained their existing prices. On the other hand, the price of a number of drugs that were unable to demonstrate bioequivalence was cut, leveling the weighted average price of the same ingredient drugs (substitutes) downward. The higher the price of the drug in negotiations as compared to the weighted average price of its substitutes, the greater the price reduction, putting the bioequivalent drugs at a relative negotiating disadvantage. According to industry sources on the 11th, the price difference between the weighted average price of the negotiated drug and the weighted average price of the equivalent drugs became larger and is expected to increase the price reduction of the respective drugs during PVA negotiations. According to the government’s Detailed Operating Guidelines for PVA Negotiations, the weighted average price of substitute drugs is one of the considerations used to adjust the reference price used in negotiations. According to Article 11 of the guidelines, considerations for adjusting the negotiated reference price include: ▲ the impact of the increase in claims of the negotiated drug has on the reduction or increase in insurance finances ▲ the impact of the negotiated drug on the weighted average price of the entire market, which includes substitute drugs. The substitute drugs for ‘Type C’ PVA are drugs with the same ingredient and route of administration as the negotiated drug. As the weighted average price of same-ingredient drugs has been leveled downward due to price ceiling reevaluations, their impact on the weighted average price of negotiated drugs whose prices have been maintained by demonstrating biological equivalence is bound to increase. "As the weighted average price of substitute drugs has been leveled downward, the bioequivalent drugs may be regarded to have an adverse effect on national health insruance finances, resulting in been larger price cuts than that made using the reference formula during PVA negotiations.” said a pharmaceutical industry representative. In other words, the greater the price difference is between the negotiated drug and the weighted average price of its substitutes, the greater the price reduction may become with increased usage. The generic drugs that have demonstrated bioequivalence will be losing out because their price was maintained during the previous negotiations, rendering the price difference greater compared with the weighted average price of its substitutes, which were leveled downward. The pharmaceutical industry has been expressing frustration as the bioequivalence tests they conducted to comply with government policy are being used against them. The insurance price ceiling reevaluations on generics took place last year and this year to improve the quality of generic drugs. The government pulled out the bioequivalence card to improve quality after a number of generic products were found to contain NDMA and other carcinogens in their ingredients, including the hypertension drug valsartan. Generics that have proven their bioequivalence were allowed to maintain their prices, while generics that were easily approved through contract manufacture had their prices reduced. The government’s intention at the time was to save only those drugs that took the time and expense to prove their viability from penalties, but instead, the bioequivalent drugs are being penalized through the post-marketing control system. In response, the industry is calling on the government to find a way to improve the system so that those who cooperated with government policy are not disadvantaged during PVA negotiations. For example, the government plans to negotiate PVA drugs to reflect the contribution of items that increased production or were used to treat infectious diseases through information cooperation to address the unstable drug supply and demand issue last year. The pharmaceutical industry believes that the PVA guidelines should also reflect the drugs’ contribution to improving drug quality. "The more the items that have not demonstrated bioequivalence there are, the lower the weighted average price of substitute drugs, and the more expensive the bioequivalent drugs may appear to be, resulting in a larger negotiated price cut," said an industry official. "It is unreasonable to try to further cut drug prices just because they are relatively expensive during PVA negotiations, even though they are being well sold for abiding with government policy."

- Company
- "Enhertu gets expanded indication to treat lung cancer"
- by Son, Hyung-Min Jul 11, 2024 06:12am
- Ahn Myung Ju, Professor of the Division of Hematology-oncology in the Department of Medicine at Samsung Medical Center “Previously, patients could only use Enhertu by participating in clinical trials conducted in tertiary general hospitals. Current approval in South Korea will provide a new treatment option for patients with HER2 mutant metastatic non-small cell lung cancer (NSCLC). Brain metastasis is common in lung cancer patients. Because Enhertu demonstrated a response rate of 50% in patients with brain metastasis, it is clinically significant.” During a recent meeting with Daily Pharm, Ahn Myung Ju, Professor of the Division of Hematology-oncology in the Department of Medicine at Samsung Medical Center, highly regarded the clinical value of Enhertu (ingredient: trastuzumab deruxtecan). Enhertu, an antibody-drug conjugate (ADC) jointly developed by Daiichi Sankyo and AstraZeneca, has been approved for the treatment of HER2-positive breast cancer and gastric cancer. As Enhertu demonstrated to be effective in HER2 mutant NSCLC and HER2-low breast cancer, its indication was expanded in May. HER2 mutant NSCLC is a rare cancer that occurs in about 2-4% of patients with NSCLC. Previously, the treatment of this cancer with chemotherapy and immunotherapy for cancer was limited, and HER2-targeted therapy showed inconsistent results. Therefore, there has been an unmet need for an effective treatment option to increase the survival in patients. Previously, several targeted treatment options, such as Hanmi Pharm’s poziotinib, were tested for the treatment of HER2 mutant NSCLC but failed to demonstrate efficacy. Therefore, Enhertu is likely to provide new hope to patients with HER2 NSCLC, which is categorized as a rare cancer. Ahn said HER 2 mutation gained attention in NSCLC after the availability of next-generation sequencing (NGS) testing and targeted therapies like Enhertu. It will provide a new treatment opportunity to patients with NSCLC. Enhertu is approved in South Korea based on phase 2 trials Enhertu is an ADC that targets HER2 mutations by linking an antibody to a payload that destroys tumor cells. This treatment works by binding antibodies to specific antigens, triggering an Antibody-Dependent Cellular Cytotoxicity (ADCC) reaction, and allowing the payload to enter tumor cells and exit through their membranes. This mechanism results in a bystander effect, enabling the destruction of cancer cells, including those without HER2 mutations, demonstrating its anti-tumor efficacy. Enhertu is effective in various diseases, including breast cancer, gastric cancer, colorectal cancer, and NSCLC. In DESTINY-Lung02 study, Enhertu demonstrated antitumor response to the second-line treatment of HER2 mutant metastatic NSCLC. This study evaluated the efficacy and safety in patients with advanced, unresectable, or metastatic NSCLC who have previously been treated with systemic therapy, including platinum-based chemotherapy, more than once. The clinical results demonstrated that Enhertu had confirmed ORR of 49%, complete response (CR) of 1%, and partial response (PR) of 48%. The mean duration of response (DOR) was 16.8 months. “Previously, the second-line treatment, docetaxel, showed a response rate of only 10-15% and a median progression-free survival (PFS) of just 5-6 months. In contrast, Enhertu showed a response rate of 50% and a durable response period lasting up to 16 months. Moreover, brain metastasis is commonly found in lung cancer, and Enhertu has demonstrated an intracranial objective response rate (IC-cORR) of over 50%, even in patients with brain metastasis,“ Ahn said. “Kadcyla, previously used for HER2-positive breast cancer, showed a response rate of over 50% in non-small cell lung cancer (NSCLC) but had limitations due to the short duration of response. In this context, Enhertu has recently gained attention for demonstrating efficacy in HER2-mutated metastatic NSCLC patients,“ Ahn said. ”Although it’s a phase 2 single-arm study, Enhertu demonstrated better outcomes than data from existing medications, warranting consideration for reimbursement,“ Ahn added. "Enhertu use will be greater" in HER2 NSCLC In the case of metastatic non-small cell lung cancer (NSCLC) with HER2 mutation, it represents about 2-4% of all NSCLC cases.This cancer type is characterized by adenocarcinoma histology, prevalence in non-smokers and females, and a higher incidence among East Asian populations, including Japan and Korea. Additionally, it exhibits a higher rate of brain metastases. HER2 abnormalities are categorized into ‘gene mutations,‘ ‘gene amplifications,‘ and ‘protein overexpression.‘ In most cancers, such as gastric cancer and breast cancer, HER2 abnormalities mainly manifest as an overexpression of the HER2 protein. Previously, HER2 overexpression gene abnormalities were generally considered to not play a significant role in NSCLC. First-generation platinum-based chemotherapy has traditionally been the standard treatment for HER2 mutant non-small cell lung cancer. Recently, combination therapies of platinum-based chemotherapy and immunotherapy for cancer have also been employed. However, non-smokers are known to have a poorer response to immunotherapy for cancer, leading to worse prognosis. “So far, in HER2-mutated metastatic non-small cell lung cancer (NSCLC), drugs like Giotrif (afatinib), which is developed to target Epidermal Growth Factor Receptor (EGFR) mutations, have been used, but their response rates have been low, around 10%,“ Ahn said. “Additionally, poziotinib showed a better response rate of approximately 27%, but its use has been challenging due to significant toxicity and side effects,“ Ahn added. Ahn said that Enhertu will bring many changes to the treatment of NSCLC. “In the United States, Enhertu is indicated for the treatment of HER2 mutant metastatic NSCLC with the U.S. FDA approval, and it is widely used with reimbursement coverage. Even in Europe with stringent standards, non-small cell lung cancer patients are strongly recommended to undergo HER2 mutation testing, and those with HER2 mutations are advised to use Enhertu,“ Ahn said. “It is unfortunate that patients cannot afford the monthly cost of approximately 7 to 8 million won for Enhertu, which allows them to maintain their daily lives for over a year. Therefore, it would be beneficial if the government explores various measures, such as selective reimbursement, to alleviate the financial burden on patients, even if it means slightly increasing the portion they pay out of pocket,“ Ahn emphasized.
- Company
- Will Trodelvy be deliberated for reimb by DREC in August?
- by Eo, Yun-Ho Jul 11, 2024 05:47am
- The road to reimbursement for the new ADC breast cancer drug Troldelvy remains a bumpy one. The agenda remains pending for 8 months now. Gilead Sciences' triple-negative breast cancer (TNBC) drug Troldelvy, whose reimbursement request received 100,000 consents in a public petition, was not presented for deliberation to the Health Insurance Review and Assessment Service's Drug Reimbursement Evaluation Committee in July. The drug’s application has remained without progress since its reimbursement standard was set by the Cancer Disease Review Committee in November last year. Therefore, attention is now focused on whether the drug will be submitted to DREC in August. Trodelvy is already listed in about 30 countries around the world. Taiwan, which has a single-payer healthcare system similar to South Korea's, began reimbursing Trodelvy in February this year. The global rush to improve patient access to Trodelvy has been driven by the poor treatment environment for metastatic triple-negative breast cancer and the clinical value of Trodelvy. Triple-negative breast cancer is an aggressive form of breast cancer that recurs and metastasizes rapidly. Patients with metastatic triple-negative breast cancer who have metastasized despite treatment have a life expectancy of only a few months even with chemotherapy. However, chemotherapy has long been the standard of care due to the lack of targets that can effectively kill cancer cells. Trodelvy, the first Trop-2-targeted antibody-drug conjugate (ADC), is the only treatment for metastatic triple-negative breast cancer in the second-line or higher setting that has been shown to prolong survival compared to chemotherapy and has settled as the global standard of care since its introduction. Currently, major guidelines in the U.S. and Europe specify Troldelvy as the preferred agent for patients with previously treated metastatic triple-negative breast cancer. In a Phase III study, the overall survival of the chemotherapy arm was 6.9 months, compared to a nearly one-year survival. (11.8 months) in the Troldelvy arm, In addition, Troldelvy demonstrated an effect in controlling symptoms and pain caused by cancer and an improvement in patients' quality of life by improving their overall health status. Trodelvy was awarded the highest possible score of 5 points on ESMO-MCBS, the European Society for Medical Oncology's (ESMO) scale used to rate the value of anticancer drugs. A score of 5 indicates that a drug is effective not only in prolonging patient survival but also in improving quality of life, and Troldelviy is the only treatment for metastatic triple-negative breast cancer to receive a score of 5 on ESMO-MCBS. In fact, the U.K. has detailed the rationale behind its assessment, stating that the state’s reimbursement decision was based on the severity of metastatic triple-negative breast cancer and the survival benefit of Troldelvy. Similar to Korea, the U.K. uses the incremental cost-effectiveness ratio (ICER) to evaluate new drugs for health insurance coverage. While the UK has one of the highest reimbursement barriers for new drugs, it applies flexible pharmacoeconomic evaluation criteria for innovative drugs used for serious conditions to improve patient access. In the UK, Troldelvy was granted preferential economic evaluation because it prolonged survival in terminally ill patients with less than 2 years of life expectancy, whose population is even smaller than those of rare diseases. As a result, Trodelvy gained access with an ICER threshold that was approximately twice higher than that of general drugs. Meanwhile, Troldelvy has been the subject of a series of petitions this year, gathering more than 100,000 consents online. Since the petition system’s inception, Trodelvy is the only drug to be referred to the National Assembly for garnering more than 50,000 signatures. The Korean Alliance of Patients' Organizations also responded to the desperate pleas of patients and their caregivers when the petition was abandoned due to the expiration of the 21st National Assembly's term. In May, the organization submitted a letter directly to the Ministry of Health and Welfare requesting a prompt review of the reimbursement of drugs with high patient demand, including Trodelvy.
- Policy
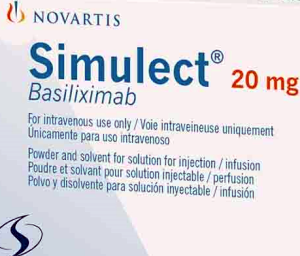
- Simulect in supply crisis with no alternative
- by Lee, Tak-Sun Jul 11, 2024 05:47am
- Simulect Inj (basiliximab, Novartis Korea), which is used as an induction therapy to prevent immune rejection in organ transplant patients, is expected to run out of supply in Korea due to issues in securing the supply the drug’s active pharmaceutical ingredient. In particular, there is no alternative drug for liver transplantation, increasing the need for the government’s prompt response. Novartis Korea reported to the Ministry of Food and Drug Safety on the 8th that the company expects a supply shortage due to the lack of global supply of the API for Simulect Inj. Due to the API issue, the amount scheduled to be imported in September will not arrive. As a result, the company expects the remaining stock to run out by mid-January next year based on the current inventory. Simulect is a leading induction therapy drug used to prevent immune rejection during organ transplantation. It is reimbursed for kidney, heart, liver, lung, small bowel, pancreas, and pancreatic islet transplants. The company noted that while hospitals and transplant centers may be able to reschedule living donor transplants, which account for approximately 70% of all transplants, in the short term, a shortage of Simulect could result in changes in the physical condition of the donor and recipient during the delay, which could affect the outcome of the transplant and result in the postponement of all other scheduled surgeries. The company added in the case of the rest - the 30% deceased donor transplants - timing is critical due to the nature of the donor, so the surgery must be performed quickly, and as surgery is nearly impossible without induction therapy, both the organ donation and the surgery may be rendered impossible. The problem is liver transplants. There are currently no other approved or available drugs for induction therapy for liver transplants, although around 1,500 cases are performed per year. In the worst-case scenario, liver transplants could become unavailable. Novartis plans to accelerate the approval of its new finished product manufacturing site. By the end of August, it could begin shipping products manufactured in Italy. However, domestic approval has not been granted for imports from that plant yet. "As the new manufacturing site can be put into producing finished products upon approval of the plant change request, we will work to minimize the supply shortage period through various measures, including efforts to accelerate the approval of the request.” In March, Simulect was also hit with a three-month import suspension when it recalled products due to possible glass particle contamination in the attached ampoules. However, the company responded quickly and did not disrupt supply. However, as this is a global API supply issue, if the administrative process of replacing the manufacturing plant is delayed, domestic supply and demand instability is deemed inevitable. This is why appropriate support from regulatory agencies such as the Ministry of Food and Drug Safety is necessary.

- Company
- From obesity to brain diseases...expansion of GLP-1 agonists
- by Son, Hyung-Min Jul 11, 2024 05:46am
- In-Young Choi, Head of R&D at Hanmi Pharmaceutical The domestic pharmaceutical bio industry is looking into the possibility of developing various new drugs with GLP-1 agents. As Novo Nordisk and Lilly's GLP-1-based obesity drugs have become global blockbusters, latecomers are also avidly developing jumping in to develop the next blockbuster. Major domestic companies are conducting clinical studies on the use of GLP-1 agents not only for obesity, focusing on the mode of administration and quality of weight loss effects, but also in the field of degenerative brain diseases such as metabolism-associated steatohepatitis (MASH) and Alzheimer's disease. On July 10, the Korea Biotechnology Industry Association and RX Korea hosted the BIOPLUS-INTERPHEX KOREA 2024 (BIX 2024). At the event, major Korean pharma and biotech companies introduced their GLP-1 drug candidates. GLP-1 drugs can be effective in weight loss by increasing satiety and improving insulin secretion and sensitivity, resulting in favorable glycemic control. Recently, GLP-1 agents have been reported to be effective against cardiovascular diseases and kidney diseases, and various clinical studies are underway on their use in patients with alcoholism and dementia. At the event, Hanmi Pharmaceutical introduced its new drug candidate for obesity which has a novel mode of action. The company is developing HM15275, an obesity drug candidate that simultaneously acts on glucagon-like peptide-1 (GLP-1), glucose-dependent insulinotropic peptide (GIP), and glucagon. HM15275 recently entered a Phase 1 clinical trial in the U.S. To date, a dual GLP-1-GIP receptor agonist Zepbound has been released, but there is no commercialized triple agonist that also includes glucagon. Hanmi Pharmaceutical is aiming to launch a first-in-class drug in this category. In preclinical trials, HM15275 showed less muscle mass loss and higher weight loss than Lilly's obesity drug tirzepatide (Zepbound). In-Young Choi, Head of R&D at Hanmi Pharmaceutical, said, "There are three strategies to developing a third-generation obesity drug following Wegovy and Zepbound. The strategies include increasing the dose, targeting a new mode of action (MOA), or adding substances such as glucagon that can increase the weight loss effect. We are exploring all possibilities through our various pipelines.” Choi added, "There is still an unmet demand for the weight loss effects of GLP-1 obesity drugs such as Zepbound and Wegovy, and the quality of weight loss and reduction of the weight rebound will be the indicators of competitiveness." Mi-Kyung Kim, Vice President and Head of Research HQ at Dong-A ST Also at the event, Mi-Kyung Kim, Vice President and Head of Research HQ at Dong-A ST, introduced its company’s new obesity drug DA-1726. According to Kim, The company completed preclinical studies and is in Phase I clinical trials for DA-1726., with the first patient dosing in April of this year. DA-1726 is a new drug candidate that is being developed as a long-acting oxyntomodulin peptide analog for the treatment of obesity. It acts simultaneously on both GLP-1 and glucagon receptors to suppress appetite, stimulate insulin secretion, and increase basal metabolism in the periphery, ultimately leading to weight loss and glycemic control. In preclinical trials, DA-1726 demonstrated similar weight loss efficacy despite higher food intake compared to tirzepatide, with less rebound after weight loss compared to tizetapide. Kim explained, "The U.S. Food and Drug Administration (FDA) requires a weight loss effect of 5% or more for an obesity drug, but the market is looking for more. We are focusing on the quality of weight loss." GLP-1 agents show promise in MASH·brain disorders GLP-1 drugs are also showing promise in the field of brain diseases. Recently, the mechanism of action of GLP-1 drugs that block neuroinflammatory responses by targeting microglia has been discovered, and active clinical studies are underway on their use in Parkinson's disease and Alzheimer's disease. There is already evidence that GLP-1s are associated with a reduced risk of developing dementia. A study of 88,381 patients with type 2 diabetes aged 65 years and older who were treated with liraglutide-based Victoza showed that those taking Victoza had a lower risk of dementia than those in the other treatment groups. Seulki Lee, CEO of D&D Pharmatech Among domestic companies, D&D Pharmatech is conducting clinical trials of GLP-1 agents in various areas, including Parkinson's disease and dementia. However, in 2020, D&D Pharmatech failed to demonstrate efficacy in a Phase II clinical trial involving 255 patients with Parkinson's disease. The primary endpoint, symptom improvement after a total of 36 weeks of treatment, did not show a statistically significant effect compared to placebo. A closer look revealed a significant difference between the NLY01 and placebo groups at 24 weeks post-dose. However, between weeks 24 and 36, the placebo group showed more improvement than NLY01. D&D Pharmatech is also conducting clinical studies on GLP-1 agents in MASH as well as neurodegenerative diseases. Recently, the company received the U.S. Food and Drug Administration's (FDA) IND approval to initiate a global Phase II trial for DD01, a new drug candidate for MASH. The Phase II trial will enroll 68 overweight and obese patients with MASH at 10 clinical sites in the U.S. D&D Pharmatech’s DD01 is a GLP-1-glucagon dual receptor agonist that has demonstrated weight loss and fatty liver reduction in preclinical studies. In a Phase I trial, DD01 reduced fatty liver by more than 50% at 4 weeks of treatment. Seulki Lee, CEO of D&D Pharmatech, said, "Although we did not meet the primary endpoint while developing a new drug for Parkinson's disease, we have seen potential. Our current focus is on metabolic diseases. We are looking forward to further clinical results based on the promise we have seen with GLP-1 agents in MASH."
- Policy
- Debate within the Health and Welfare Committee members
- by Lee, Jeong-Hwan Jul 10, 2024 05:48am
- Members of the Democratic Party of Korea and the ruling party who are also committee members of the Health and Welfare Committee of the National Assembly show significant differences in viewpoints regarding President Yoon Suk Yeol’s policy of expanding medical school quotas, and the current situation surrounding the conflict between the medical community and the government, and the medical crisis. The ruling party argues that the National Assembly must support the government’s medical reforms to increase the number of doctors and help essential and regional medical services. The Democratic Party of Korea argues that the government must first clearly take responsibility for the medical exodus caused by forced expansion. As the viewpoints of the ruling and opposition parties diverge on government's medical reforms, such as increasing the medical school quota, it has become increasingly difficult to establish a bipartisan deliberation committee to resolve conflicts between the medical community and the government. On July 9th, members of the People Power Party and the Democratic Party, who are committee members of the Health and Welfare Committee, held a separate press conference and criticized the other party. The Democratic Party says, "The government must admit to causing the medical community-gvt conflicts and medical crisis" During the press conference, the Democratic Party condemned the People Power Party's withdrawal from adopting a bipartisan resolution and opposing the specification of 'government responsibility during the emergency hearing on the medical crisis. They argue that the People Power Party is responsible for the 13-hour hearing without a result or resolution. The Democratic Party argued that the Presidential Special Committee on Medical Reforms is clearly limited in capacity and appealed for the need to establish a public discussion committee on medical reforms involving both parties, the government and experts. Kang Sunwoo, Secretary of the Democratic Party, said, "We are disappointed in the ruling party's refusal to take even minimal responsibility to correct the government's clear policy failures." He emphasized, "The People Power Party held a superficial hearing in Yongsan, not for the people." The People Power Party says, "Medical reforms, both parties must collaborate instead of political conflicts" In the afternoon of the same day, the People Power Party held a press conference to refute the Democratic Party's claims. The party asserted that the Democratic Party is politicizing medical reforms to improve essential healthcare and regional healthcare into a political issue. The People Power Party said that previously, the Democratic Party government had pursued increasing medical school admissions but was hindered by opposition from the medical community. They emphasized that achieving medical reforms, a challenging task, requires collaborative efforts across government administrations, regardless of political affiliation. They argued that the Democratic Party must stop arguing if it hopes to resolve the medical crisis quickly and improve essential and regional healthcare. Kim Miae, Secretary of the People Power Party, said, "Instead of addressing patient anxiety caused by trainee doctors leaving and medical staff going on strike, the Democratic Party, during a press conference, claimed that the ruling party unilaterally rejected the resolution." She added, "The Democratic Party is trying to politicize the current situation and emphasize only government responsibility as a tactic. We must put joint efforts into problem-solving rather than politicizing the issue." Meanwhile, the Health and Welfare Committee will commence a general meeting on the upcoming 16th and listen to work reports from the government departments, including the Ministry of Health and Welfare (MOHW) and Ministry of Food and Drug Safety (MFDS). During the meeting, the Democratic Party of Korea and the ruling party are expected to participate in questioning and discussion to formulate a solution to resolve the conflict between the medical community and the government and the medical crisis.