- LOGIN
- MemberShip
- 2026-07-22 18:36:19
- Company
- ADC MM drug Blenrep set for commercialization in Korea
- by Eo, Yun-Ho Nov 26, 2025 06:09am
- The new multiple myeloma drug Blenrep is expected to soon become commercially available in Korea. The Ministry of Food and Drug Safety (MFDS) is currently conducting the final review for the approval of GSK Korea’s Blenrep (belantamab mafodotin), the first anti-BCMA (B-cell maturation antigen) antibody-drug conjugate (ADC) in its class, developed by GSK Korea. Industry expects the approval could come as early as this year. Blenrep was approved in Europe last July and in the United States last October. The anticipated approved indications for Blenrep are ‘in combination with bortezomib and dexamethasone (BVd) in adult patients with relapsed or refractory multiple myeloma who have received one or more prior therapies,’ and ‘pomalidomide plus dexamethasone (BPd) in patients who have received one or more prior therapies, including lenalidomide (Revlimid).’ This drug demonstrated efficacy through the Phase III DREAMM-7 trial. In patients who had received at least 2 prior therapies, Blenrep combination therapy reduced the risk of death by 51% compared to the Darzalex (daratumumab)-based triple regimen (DVd). The median progression-free survival (PFS) was 31.3 months for the Blenrep combination therapy group versus 10.4 months for the Darzalex-based triple regimen group, representing approximately a threefold improvement in the Blenrep combination therapy group. The safety and tolerability profile of the Blenrep combination was generally consistent with what is known for the individual agents. However, the Blenrep arm showed a higher incidence of ocular toxicity, such as keratopathy and vision changes. Due to this safety concern, the FDA advisory committee voted against Blenrep’s reapproval in July. Meanwhile, GSK is conducting a clinical program to demonstrate Blenrep’s potential benefit in earlier-line treatment settings. Overall survival follow-up for the DREAMM-7 and DREAMM-8 trials, which include patients who received at least one prior line of therapy, is ongoing. Results from these studies are expected to be released in 2028.
- Company
- Bispecific antibody opens new era of DLBCL treatment
- by Son, Hyung Min Nov 26, 2025 06:09am
- The treatment landscape for diffuse large B-cell lymphoma (DLBCL) is changing quickly. Roche's bispecific antibody, which was previously a third-line option, received an expanded indication for second-line treatment, broadening the therapeutic strategy for DLBCL. Given the aggressive nature of DLBCL, where prognosis rapidly worsens after first-line failure, there is significant attention on whether a more potent combination treatment and a new mechanism of action can increase patients' chances of survival. On November 25, Roche Korea held an event at its headquarters in Gangnam-gu, Seoul, to discuss the unmet needs in the DLBCL treatment landscape and the clinical value of its new drug. Roche has already launched products in this field, including Polivy (polatuzumab vedotin) and the bispecific antibody 'Columvi (glofitamab)'. Currently, Polivy can be used as a first-line treatment in combination with cyclophosphamide, doxorubicin, and prednisone (R-CHOP), and Columvi can be used for subsequent lines of treatment. In July, Columvi received an expanded indication from third-line to second-line treatment. The specific indication is for adult patients with relapsed or refractory DLBCL not otherwise specified (DLBCBL NOS) who are ineligible for autologous stem cell transplantation (ASCT), in combination with gemcitabine and oxaliplatin. Professor Seok Jin Kim of the Department of Hematology-Oncology at Samsung Medical CenterColumvi has a 2:1 structure, in which two binding domains target the CD20 surface antigen expressed on malignant B cells, and one domain targets the CD3 antigen expressed on immune T cells. This structure allows for a more robust and stable binding. The efficacy of Columvi was confirmed through the Phase 3 STARGLO study. The trial included patients with relapsed or refractory DLBCL who were ineligible for ASCT after one or more prior systemic therapies, or who had a history of two or more prior systemic therapies. The two-year follow-up results demonstrated that the Columvi + gemcitabine + oxaliplatin combination therapy reduced the risk of death by 41% compared with rituximab + gemcitabine + oxaliplatin combination therapy. The Columvi combination group's Progression-Free Survival (PFS) was 13.8 months, approximately a four-fold increase from 3.6 months in the rituximab combination group. The rate of patients achieving Complete Response (CR) was higher in the Columvi combination group (58.5%) than the control group (25.3%). Professor Seok Jin Kim (President of the Korean Society of Hematology) of the Department of Hematology-Oncology at Samsung Medical Center commented, "While remission can be reached, maintaining it is not easy. Patients in the Columvi combination group survived without disease progression for even one year after the end of treatment. This is a significant finding." Still high unmet needs in DLBCL…"more treatment options must be secured" DLBCL is a disease in which B cells, which protect the body, grow or multiply uncontrollably. It is the most common subtype, accounting for about 40% of Non-Hodgkin Lymphomas. The disease is characterized by its aggressive nature, with rapid progression through stages. The number of DLBCL patients in South Korea was 14,183 as of last year, a 36% increase from 10,428 in 2018. Up to 15% of DLBCL patients fail to respond to the first-line standard treatment, and among those who achieve CR, 25% experience relapse within 18 months. Patients with relapsed/refractory DLBCL face a rapidly deteriorating prognosis as the line of treatment increases. Seunghun Lee, Medical Lead at RochePolivy combination therapy is known to be effective in about two-thirds of patients when used as first-line treatment. However, this means that approximately one-third of patients do not benefit from first-line treatment. Chimeric Antigen Receptor T-cell (CAR-T) therapies and bispecific antibodies are now available for subsequent treatment lines. Currently, second-line treatment options include Gilead's CAR-T drug 'Yescarta (axicabtagene ciloleucel)' and the antibody drug Columvi. For the third line, Novartis's ''Kymriah (tisagenlecleucel)' and AbbVie's 'Epkinly (epcoritamab)' are utilized. However, all options except Kymriah are currently non-reimbursed options. Professor Kim stressed, "CAR-T and bispecific antibodies are not to be compared directly. Patients who can tolerate the side effects choose CAR-T, and those who cannot choose the bispecific antibody. If all new drugs were reimbursed, prescribing would align with global guidelines. It is not appropriate to evaluate which drug is superior," pointing out, "The problem is that even when effective treatment options exist, patients are forced to repeat the same treatment due to regulatory approval and reimbursement hurdles." Seunghun Lee, Medical Lead at Roche, said, "The reason the U.S. FDA rejected Columvi for second-line approval was that it did not meet the criteria for the number of enrolled patients, but since it has been approved in major countries in Europe and Asia, we believe the racial differences are not significant," and emphasized, "Roche is currently strengthening its leadership by launching a variety of treatments for hematologic malignancies. We will continue to expand the range of choices in the DLBCL area to enable personalized treatment strategies for different patient groups."
- Company
- Bio investments hit 3yr high...investor sentiment recovers
- by Cha, Jihyun Nov 26, 2025 06:09am
- Venture capital (VC) new investments in the biotech and medical sectors during the third quarter of this year reached their highest level in 3 years, both in scale and proportion. Quarterly investment volume increased by 56% compared to the previous quarter, and the bio/medical VC investment share within the industry rose to 20%. This is interpreted as a clear sign of recovery of the once-frozen biotech investment market. According to the Korea Venture Capital Association on the 25th, new investments in the bio-medical sector during the third quarter totaled at KRW 396.9 billion. This is a sharp 56% increase from the previous quarter and a 20% rise compared to the same period last year. While overall new VC investments expanded by 33% quarter-on-quarter during the same period, the bio/medical sector's investment amount showed a much steeper growth trajectory, effectively driving the market’s rebound. The trend of increased investment in the bio-medical sector is also evident in the share indicators. The bio/medical sector accounted for 20% of all new VC investments in Q3, up 3 percentage points from the previous quarter. The fact that the share of bio-medical investments expanded despite the overall new investment pie growing larger quarter-on-quarter indicates a strong shift in capital toward biotechnology over other industries. As a result, new investments in the bio/medical sector this quarter reached their highest level in the past 11 quarters, both in absolute investment and share. This signals a full revival of previously subdued investment sentiment, with analysts suggesting the bio/medical investment trend has moved beyond a short-term rebound and entered a phase of structural recovery. (Source: Korea Venture Capital Association) Investment in the biotech/medical sector peaked at KRW 1.677 trillion in 2021 during the COVID-19 pandemic and has been on a steep decline since. Affected by interest rate hikes and economic uncertainty, investment fell to KRW 1.1058 trillion in 2022 and further declined to KRW 884.4 billion in 2023, marking two consecutive years of contraction. The bio investment market began showing signs of a rebound last year. New investment in the bio/medical sector in 2024 reached KRW 1.0695 trillion, a 20% increase year-on-year, marking a shift in the trend. Looking at quarterly figures, the market's recovery became clearly evident last year when KRW 331.6 billion poured in during the third quarter. This recovery became even more pronounced this year. Quarterly investments have continued to rise, clearly showing that the market is in a full restoration phase. Particularly significant is this third quarter's investment volume, which significantly surpassed the record set in Q3 2024 that drove last year's rebound, but also represents the largest quarterly figure since the downturn began in 2023. The biotech industry attributes this surge in investment to: ▲ the completion of valuation adjustments, ▲ an increase in companies delivering clinical results, and ▲ expectations of interest rate cuts. Specifically, it is analyzed that VCs, who maintained a conservative stance until the first half of the year, began betting on ‘high-quality companies’ with solid technological capabilities in the second half, driven by pressure to deplete their dry powder (uncommitted investment funds). Some view the first-quarter investment cliff as having actually heightened valuation attractiveness, creating bargain-buying opportunities for companies with solid technology foundations. Looking ahead, projections suggest VC funding will enter a more pronounced phase of ‘separating the wheat from the chaff,’ focusing squarely on technological prowess. Unlike the past when investments were based solely on growth potential, the trend of capital concentrating on companies with verifiable substance—such as clinical data and commercialization feasibility—is expected to continue for the foreseeable future.
- Company
- Biogen attempts Qalsody reimb listing in Korea
- by Eo, Yun-Ho Nov 25, 2025 06:14am
- The new ALS drug Qalsody (tofersen) has begun the process of applying for reimbursement coverage in Korea. Since receiving domestic approval in August, the company has been moving rapidly to seek its reimbursement. According to industry sources, Biogen Korea’s Qalsody, a treatment for amyotrophic lateral sclerosis (ALS) associated with SOD1 (Superoxide Dismutase 1) gene mutation, is scheduled to be reviewed by the Drug Reimbursement Standard Subcommittee. Before approval, Qalsody was designated a GIFT (Global Innovative Products on Fast Track) item and as an orphan drug by the Ministry of Food and Drug Safety/ However, the road ahead is expected to be rocky due to its extremely high price. In the U.S., Qalsody costs USD 15,097 per vial (approx. KRW 22.03 million), and the annual treatment cost reaches USD 211,358 (approx. KRW 308.5 million). Biogen submitted a reimbursement application immediately after approval in August, but faced its first hurdle when reviewers requested additional data. It now awaits the Reimbursement Standards Subcommittee review on the 28th. Should the subcommittee require further review or additional data, the reimbursement schedule could face further delays due to re-review. ALS, particularly SOD1 gene mutation ALS, falls into the category of ultra-rare diseases, with fewer than 100 patients worldwide. Qalsody is the first targeted therapy developed specifically for this patient group. Because ALS presents widely variable symptom onset patterns among patients, clinical trial design and endpoint determination are extremely challenging. Experts note that the emergence of the first treatment option showing potential for improving disease progression, even in just one subset of patients, holds significant meaning in ALS, a field where drug development is exceptionally difficult. Given the U.S. Trump administration’s MFN drug-pricing stance and Korea’s historically restrictive approach to rare-disease reimbursement, experts warn that if reimbursement criteria become too narrow, the possibility of the drug's domestic launch being canceled cannot be ruled out. It remains to be seen whether Qalsody will become a reimbursed treatment option in an area with virtually no therapeutic alternatives. Meanwhile, in the Phase III VALOR study, Qalsody did not meet the primary endpoint, the ALSFRS-R functional score. However, it demonstrated reductions in the secondary endpoints: a 26-38% decrease in total SOD1 protein cerebrospinal fluid and a 48-67% decrease in plasma neurofilament light (Nfl) concentration.
- Policy
- Legalizing order for manufacturer to supply essential drugs
- by Lee, Jeong-Hwan Nov 25, 2025 06:13am
- A bill has been proposed in the Korean National Assembly to stipulate the Government Supply System in the Pharmaceutical Affairs Act to ensure the stable supply of National Essential Medicines, which are frequently in short supply due to low profitability. The bill also includes an article expanding treatment opportunities for patients using essential medicines by entrusting the practical work of ensuring a stable supply to the Korea Orphan & Essential Drug Center. On November 24, Democratic Party of Korea Rep. Young-seok Seo announced that he had officially proposed this amendment to the Pharmaceutical Affairs Act. Rep. Seo stated that the number of discontinued products has recently been increasing, primarily in essential medicines with low commercial viability. He noted that the government is currently operating a Government Supply System, such as supporting drug production through agreements with pharmaceutical companies or directly importing overseas pharmaceuticals, when supply is halted due to a company's product withdrawal. However, Rep. Seo expressed concern that the current law does not explicitly provide a legal basis for the Government Supply System, limiting its application and expansion. He further pointed out that, concerning this issue, the legal basis for supporting the resumption of production of medicines not supplied domestically is also inadequate. Therefore, Rep. Seo introduced a bill that legally mandates the Government Supply System for essential medicines and entrusts its practical implementation to the Korea Orphan & Essential Drug Center. In detail, the bill allows the Minister of Food and Drug Safety (MFDS) to mandate a pharmaceutical manufacturer to manufacture National Essential Medicines for products not currently manufactured or imported into Korea when a stable domestic supply is necessary. This bill is to be newly established under Article 41-2, titled 'Manufacture and Supply of National Essential Medicine,' of the Pharmaceutical Affairs Act. In such cases, the MFDS Minister may prepay all or part of the manufacturing costs to the relevant pharmaceutical company. Furthermore, the MFDS Minister can provide administrative and technical support to ensure a swift review process when the company applies for a product license. The MFDS Minister is also mandated to entrust the duties of manufacturing, storage, and supply of National Essential Medicines to the Korea Orphan & Essential Drug Center. The proposed bill also introduces a new article, 'Article 43-2 Emergency Import of Medicines,' into the Pharmaceutical Affairs Act. This new bill allows the MFDS Minister to supply medicines domestically through methods such as importation and provide related information to patients when an urgent domestic supply is deemed necessary or when the head of a relevant central administrative agency or related organizations and groups requests the supply. Rep. Seo explained, "This bill legally specifies the government system for the stable supply of essential medicines and entrusts the practical execution to the Korea Orphan & Essential Drug Center," and added, "It will guarantee treatment opportunities for patients requiring essential medications."

- Company
- "Disparity btwn innovation speed and policy… Economic Eval"
- by Son, Hyung Min Nov 24, 2025 06:19am
- Forum As the South Korean positive list system for pharmaceutical reimbursement marks its 20th anniversary, discussion has begun on how to reflect the value of innovative new drugs within the existing economic evaluation system. With the acceleration of technological development and the surge of high-cost innovative treatments for ultra-rare diseases, critics argue that the current framework, which focuses on cost-effectiveness, can no longer sustain the balance between patient access and financial sustainability. At the forum, '20 Years of the Positive List System for Pharmaceutical Reimbursement: Value-Based System Where Innovation Meets Regulation,' held in the National Assembly on November 21, academia, government, industry, and the media all agreed on the necessity of 'modernization of economic evaluation.' The event was jointly hosted by Democratic Party Rep. Seo Young-seok and Rep. Kim Yoon, and Reform Party Rep. Lee Joo-young, and organized by the ISPOR Korea Chapter. The system focused on cost-effectiveness limits evaluation of innovative new drugs Korea's economic evaluation system was introduced in 2006 with the government's announcement of the Drug Expenditure Rationalization Plan (DERP). The PLS, introduced the following year, established the principle that all drugs must be jointly verified for clinical usefulness and economic value through the economic evaluation process, with only those drugs proven to be cost-effective included in the reimbursement list. Although the Economic Evaluation Exemption track was established in 2015 to provide an exceptional pathway, the principle that cost-effectiveness proof is mandatory for new drug reimbursement has persisted for 20 years. However, the industry points out that this cost-effectiveness-centric structure clashes with the rapidly evolving paradigm of innovative new drugs, creating a growing gap between regulation and technology that cannot be ignored. BaeChan Kim, Unit Head at Boehringer Ingelheim Korea, directly criticized the current economic evaluation framework, stating that it has failed to keep pace with changes in treatment paradigms over the last decade. Kim said, "The problem is that as the speed of innovation has accelerated, the complexity of clinical evidence has increased exponentially," and added, "The expansion of areas where standard Randomized Controlled Trials (RCTs) are practically impossible, such as ultra-rare disease treatments, has led to an increase in innovative new drugs approved based only on single-group trials. However, the current economic evaluation and reimbursement system is still aligned with the RCT standard from 20 years ago. Economic evaluation requires evolution, not reinforcement." Kim highlighted Korea's position by citing international comparisons. According to data from the U.S. Pharmaceutical Research and Manufacturers of America (PhRMA), analyzing reimbursement rates for innovative new drugs across nine developed countries, Australia, Korea, and Canada fall behind significantly, with reimbursement rates about half the average. Kim pointed out, "It is a contradictory structure where countries called 'innovation leaders' are actually the furthest behind in access to innovative new drugs." There was also criticism that limiting economic evaluation to a cost-effectiveness-centric structure is no longer realistic. Lee Sang-il, Head of the Department of Policy Planning at the National Evidence-based Healthcare Collaborating Agency (NECA), assessed, "The challenges of securing both patient access and financial sustainability have become incomparably difficult compared to the past, due to accelerated aging, the emergence of ultra-high-cost innovative drugs, and increased uncertainty in clinical evidence," and said, "We are in an era where cost-effectiveness alone cannot explain even half of the decision-making process." Lee particularly cited the absence of entry evaluations and the failure to establish a post-market evaluation system as the biggest structural problems. Lee said, "For high-cost drugs where there is a disparity between RCT data and real-world clinical data, the insurer needs to conduct a health-economic evaluation directly or through objective verification by a third party. The sustainability of drugs entering via the Risk-Sharing Agreement (RSA) or economic evaluation exemption tracks cannot be guaranteed without post-market management." He emphasized the need to establish a Korean-style Managed Entry Agreement (MEA) that defines the post-market evaluation system concurrently with the listing decision. The event was jointly hosted by Democratic Party Rep. Seo Young-seok and Rep. Kim Yoon, and Reform Party Rep. Lee Joo-young, and organized by the ISPOR Korea Chapter. It has been confirmed that such a problem exists not only in the industry but also in the media covering the regulatory sector. This suggests that the difficulties in the field are repeatedly exposed in official discussions and in practical, real-world cases. Yun-Ho Eo, a journalist at DailyPharm, pointed out, "The core message from the field is that the reason for the increase in economic evaluation exemptions is not that companies want the exemption, but that the current health-economic evaluation itself has become impossibly difficult to handle." Eo added, "Companies are mobilizing law firms, economic evaluation expert agencies, and external consultants because they cannot manage the process using only in-house personnel," and stated, "This shows that Korea's economic evaluation procedures are extremely complicated and rigid on a global scale. Korea's low drug prices and slow listing speed are recognized as a policy reality by the industry." Agrees on modernizing the economic evaluation system...Has proposed directions to strengthen review and evaluation In response to concerns raised by industry and academia, the government acknowledged the limitations of the existing economic evaluation framework and agreed on the need for institutional improvement. However, it clarified that what is needed is not the reinforcement of the system, but its refined modernization. The government highlighted securing evaluation flexibility and expertise, and establishing a post-market management system that reflects actual clinical outcomes after listing, as the core directions for reform. (from left) Sook-hyun Lee, Head of HIRAYeon-sook Kim, Director of Pharmaceutical Management Division at MOHW, said, "Korea was the first country in Asia to adopt the economic evaluation system and has steadily expanded access through institutional evolution, including the Risk-Sharing Agreement (RSA) and Economic Evaluation Exemption." Kim noted, "However, the pace of change recently is fundamentally different from when the system was designed." Kim said, "In an era where innovative new drugs based on uncertainty are surging, the current evaluation method is no longer manageable. I realized the limitations firsthand after working in the field for five months," and added, "We cannot be satisfied with the current system." Kim then presented two tasks: first, the need to define 'innovativeness' more clearly and precisely in the guidelines adopted by HIRA; and second, the need to establish a structure that systematically evaluates actual clinical outcomes after listing and feeds that back into reimbursement. Kim said, "Post-market evaluation is fundamental to all healthcare programs, but it is not yet fully established in Korea. We must create a structure that confirms actual efficacy while minimizing the burden on the clinical and industrial sectors," and added, "Our system lacks flexibility and expertise compared to those of developed countries." We will develop the system in the most practical way possible." The Health Insurance Review & Assessment Service (HIRA) also acknowledged the conflict between technological innovation and the existing evaluation framework, stating that an adjustment to the evaluation structure is unavoidable. Sook-hyun Lee, Head of HIRA's New DRG Review Division, said, "Ultra-high-cost innovative new drugs like cell and gene therapies, ADCs, and one-shot treatments are entering the actual listing stage," and added, "The question is whether the cost-effectiveness evaluation alone is sufficient to determine the appropriate price for these drugs." Lee stated, "We cannot process all drugs through the economic evaluation system, nor can we exempt all drugs from it," explaining the currently operating hybrid management system. She emphasized that various forms of conditional reimbursement models have been introduced, including the refund-type RSA, the pre-listing/post-evaluation model, and the requirement for post-listing submission of clinical data. Regarding the recent increase in economic evaluation exemptions, Lee stressed that this should be understood not as a hurdle-free passage, but as a conditional system intended to expand access. Lee explained, "The uncertainty of ultra-high-cost/ultra-rare drugs cannot be resolved simply by increasing the ICER threshold or reinforcing the economic evaluation system," and concluded, "Given the variance in RWD quality, the gap in evidence levels, and the difficulty of quantitative evaluation, a comprehensive adjustment of the evaluation structure is necessary." Lee then added, "Economic evaluation and its exemption cannot be viewed as a simple dichotomy. We will expand post-market management and value-based evaluation to match technological changes." The government largely agrees with industry and academia on the problems and has repeatedly emphasized that the time calls for a more sophisticated evaluation method, not for strengthening regulations. The assessment is that the core task for the next 20 years will be precisely balancing the conflicting policy goals of access, sustainability, innovation, uncertainty, and pre- and post-market evaluation.
- Opinion
- [Desk View] Dual pricing for the sake of our own citizens
- by Eo, Yun-Ho Nov 24, 2025 06:19am
- The government intends to expand the number of dual-priced listed drugs to prevent disclosure of actual transaction prices. By raising reference prices through higher public list prices, it aims to improve access to new drugs. The government has announced a major overhaul of Korea’s drug pricing system. The forthcoming reform plan, which the government plans to reveal concrete details soon, is drawing significant attention from the pharmaceutical industry. It is expected to include adjustments to the generic drug pricing calculation rate, reform of the tiered drug pricing system, consolidation of post-market management systems, expansion of the Risk Sharing Agreement (RSA) and dual pricing scheme, and R&D investment-linked drug price premiums. Multinational pharmaceutical companies are particularly focused on expanding the dual pricing system. Among RSAs, the refund-type model—which assigns dual prices by separating the actual transaction price from the listed price—has consistently been the preferred contract type since its introduction. However, calls for improvements to its scope have been persistent. Many even suggested excluding the refund-type from RSAs altogether. The government has also partially accommodated these opinions, making minor adjustments to the system. Initially applied under a strict criterion, only to ‘anticancer drugs or rare disease treatments with no equivalent substitutes or therapeutically equivalent alternatives’, the ‘life-threatening’ qualifier has now successfully been removed from the criteria. Yet, the prevailing sentiment remains that its application conditions are still stringent. However, this time appears to be different. Pressure from the U.S. Trump administration’s Most-Favored-Nation (MFN) drug pricing policy served as a trigger, and the health authorities appear to have internalized growing alarm over the long-discussed but uncomfortable issue of “Korea passing” within the global pharma industry. Given the circumstances, the upcoming expansion of RSA and dual pricing will likely loosen at least the criteria tied to disease severity. Whether the current price cap, “below the A7 adjusted average price,” will be modified is another point to watch. It is never right for a game of brinkmanship to unfold over reference prices, leading to the bypassing phenomenon. The products sold by multinational pharmaceutical companies are not luxury goods like Mercedes-Benz or Chanel. It is also true that drug prices are set higher in relatively poorer countries—those with weaker negotiating power. Still, many countries are expanding the share of non-public drug pricing—a second-best solution, though not ideal— to secure access to new therapies for their own citizens. While the global moral imperative of ‘transparent drug pricing’ is commendable, the government must also make decisive choices for the sake of patients in our country.
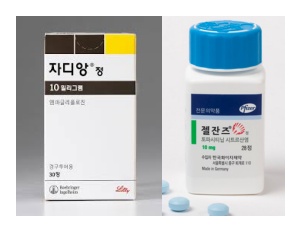
- Policy
- Price of Jardiance, Xeljanz cut 30% with entry of generics
- by Lee, Tak-Sun Nov 24, 2025 06:19am
- The diabetes treatment Jardiance (empagliflozin, Boehringer Ingelheim) and the rheumatoid arthritis treatment Xeljanz (tofacitinib citrate, Pfizer) will have their maximum insurance prices officially reduced due to the listing of generic versions. When a therapeutically equivalent product is newly listed, the price ceiling is adjusted to 53.55%, and a one-year 70% add-on premium is applied. As a result, starting in December, the previous prices will drop by 30 percentage points. According to industry sources on the 21st, starting December, the price of Xeljanz 5mg Tab will decrease from KRW 10,996 to KRW 7,697, and Xeljanz 10mg Tab will decrease from KRW 18,250 to KRW 12,775. Additionally, the price of Jardiance 10mg Tab will be adjusted from KRW 582 to KRW 408, and Jardiance 25mg Tab from KRW 762 to KRW 533. This price adjustment is due to the inclusion of generic drugs in the reimbursement list. With the substance patent for Xeljanz expiring on the 22nd of this month, 17 products from 12 companies, including those from Daewoong Pharmaceutical and Chong Kun Dang, will be reimbursed starting on the 23rd. Generic versions of Jardiance also entered the market en masse in October. Following the expiration of the substance patent, 235 products from 37 companies were listed for reimbursement on October 24. Both products are achieving high sales in the market. Jardiance recorded KRW 66.3 billion last year based on UBIST data, while the combination product Jardiance Duo recorded KRW 41.8 billion, boasting a combined market size exceeding KRW 100 billion. Xeljanz also secured market presence with KRW 14.3 billion in outpatient prescriptions. The ex officio price reductions made for the extended-release and combination formulations are expected to cause significant concern for Pfizer and Boehringer. Starting December, the price ceiling of Xeljanz XR 11mg will drop from KRW 22,169 to KRW 17,071, and Xeljanz Syrup 1mg/mL will decrease from KRW 527,808 to KRW 369,443. Additionally, the price ceiling of Jardiance Duo Tab 5/1000mg will decrease from KRW 301 won to KRW 284, Jardiance Duo Tab 5/500mg from KRW 301 to KRW 278, and Jardiance Duo Tab 5/850mg from KRW 301 to KRW 278. Among the items subject to this ex officio adjustment, Xeljanz 5mg Tab, Xeljanz 10mg Tab, Xeljanz XR 11mg Tab, and Xeljanz syrup will see an additional price reduction on November 23 next year, once the one-year add-on premium ends, dropping to 53.55%, equal to the generic price. Jardiance 10 mg and 25 mg will undergo the same adjustment on October 24 next year, one year after generic entry. For both original manufacturers, generic competition and reimbursement price cuts will inevitably impact sales, prompting intense efforts to defend market share. Pharmacies are advised to take note of returns and settlement procedures following price changes for these frequently prescribed original products. Meanwhile, drug prices will also be adjusted for the menopausal hormone therapy drug Angelic Tab (drospirenone/estradiol, Bayer) and the antifungal agent Ambisome Inj (liposomal amphotericin B, Gilead) with the entry of their respective generics. The price of Angelic Tab will be adjusted from KRW 10,393 to KRW 5,565 starting in December. The price of Ambisome Inj will be reduced by 30% from KRW 129,392 to KRW 113,218, and after the add-on premium ends on October 1 next year, the price will drop to KRW 86,612.
- Opinion
- [Reporter’s View] K-Pharma bets its survival on new drug R&
- by Choi, Da-eun Nov 24, 2025 06:18am
- The R&D direction of major Korean pharmaceutical companies is shifting from generics to new drug development. This shift comes as the perception grows that generics, long the mainstay of domestic pharma growth, are no longer a safe bet. With tighter price regulations, intensified competition, and shrinking distribution margins, the generic ecosystem is being pushed into relentless price pressure. As the sense of crisis deepens that generics alone cannot guarantee the future, leading pharmaceutical companies are increasingly willing to bear the cost burden to secure mid-to-long-term growth engines. Hanmi Pharmaceutical is aggressively investing in R&D, raising expectations that it may be the first Korean company to launch a homegrown obesity drug. Hanmi's R&D expenditure for the third quarter of this year reached KRW 169.1 billion, a KRW 15.5 billion increase year-on-year, accounting for 15% of its sales. CKD (Chong Kun Dang) is moving away from a generic-centric model toward a bio-focused structure, expanding its pipeline from ADC (antibody-drug conjugate) oncology drugs to advanced biopharmaceuticals. It recently announced a KRW 2.2 trillion investment, making a major ‘bet’ by constructing a large-scale biopharmaceutical complex R&D center in Siheung. Its R&D expenses are on the rise - from KRW 151.2 billion in 2023, KRW 157.4 billion in 2024, to KRW 126.5 billion in the third quarter of this year. JW Pharmaceutical is also steadily increasing its annual R&D spending. Its third-quarter R&D expenses this year reached KRW 74.9 billion, a 26.9% increase compared to KRW 59.0 billion in the same period last year. It is currently conducting a Phase III multinational clinical trial in Asia for its core pipeline, the gout treatment ‘epaminurad (URC102)’. Considering the growth potential of the gout treatment market and the limitations of existing uric acid-lowering drugs, analysts view it as having significant mid-to-long-term pipeline value. Even manufacturing-oriented giants like Celltrion and Samsung Biologics are jumping into the R&D competition, expanding their own pipelines as new growth pillars. Celltrion emphasized ambitions beyond biosimilars, outlining plans to develop ADCs, multi-specific antibodies, and obesity drugs. Samsung Biologics launched Samsung Epis Holdings, a bio-investment holding company, through a spin-off. Samsung Epis Holdings plans to strengthen new drug development based on EPIS NexLab’s biopharmaceutical development platform. Behind these bold future investments by domestic companies lies a shared understanding that sustainable growth is difficult to achieve, relying solely on generic drug profits. The global M&A market, policy direction, and investment capital all operate based on ‘innovation’. This means that companies unable to break free from a domestic-focused, generic-centric structure will inevitably fall behind. Of course, strengthening R&D does not guarantee immediate success. The risk of clinical failure is significant for any company, and securing funding grows increasingly difficult. Risks clearly exist behind the banner of ‘investment for the future’. However, it is becoming increasingly clear that competitiveness in the pharmaceutical industry is shifting from price to technology. These days, the phrase “We will increase our R&D investment” appears like a motto in all pharmaceutical companies' new year business plans. To the reporter, it reads as a strategic declaration directly tied to survival. The reality that generic drugs alone cannot defend corporate value is pushing companies harder for reinvestment in R&D. As the market landscape shifts, clinging solely to safe strategies leads only to regression. The direction is becoming clearer: decisively shedding inefficient businesses and concentrating resources on next-generation drug development. This is why domestic pharmaceutical companies have chosen the difficult path of restructuring toward high-value-added focus, and why their ‘long-term’ challenge deserves close attention.
- Opinion
- [Reporter’s View] K-BIO needs strength to go the distance
- by Cha, Jihyun Nov 21, 2025 06:12am
- New drug development is a war of capital. The amount of money and time required to develop a drug candidate, push it through clinical trials, and finally reach approval is beyond imagination. It is widely known that developing a single new drug typically takes 10–15 years and costs KRW 1–2 trillion. When developing high-complexity modalities such as antibody–drug conjugates (ADCs) or bispecific antibodies, the initial R&D cost grows exponentially. This is why biotechs inevitably rely on early-stage technology out-licensing models. Without a revenue base, it is difficult to independently conduct late-stage clinical trials that require hundreds of billions to trillions of won. As a result, secure preclinical or early clinical data and quickly move to out-license the technology. For drug development biotechs, technology transfer is not a choice but a survival strategy. But recently, a shift is emerging. Rather than out-licensing technologies at early stages, some leading biotechs are now moving to carry their core pipelines into late-stage clinical trials themselves. LigaChem Bio, which strengthened its capital base after Orion Group became its largest shareholder, has declared plans to push certain ADC pipelines into late-stage clinical trials independently. ABL Bio, which signed two major licensing deals with global big pharma this year alone, has also indicated its intention to take its key pipeline into late-stage trials on its own. This trend is noteworthy as it signifies that domestic companies are moving into a phase where they directly secure control over new drug development. Under the traditional licensing model, the fate of development was effectively handed over the moment the license was transferred. If the partner changed strategy or de-prioritized the project, the original developer had no way to intervene or revive it. Conversely, by directly advancing into late-stage clinical trials, the company itself can determine the value and direction of the new drug. The decision to take a pipeline into late-stage development is also a major shift in terms of corporate value. Licensing deals grow larger the further the pipeline progresses. While early-stage technology exports typically result in contracts worth tens to hundreds of billions of won, the contract size jumps to at least hundreds of billions of won, and can reach trillions of won, by the time late-stage clinical trials commence. Behind the decision to “carry it all the way through” lies the calculation that taking on additional risk is worth it if it means the results and rewards remain with the company. Above all, the attempt to directly navigate through to the late-stage clinical phase holds significant meaning in terms of accumulating the ‘core know-how involved with new drug development’ that Korea has been lacking. Under the license-out-centric structure, most late-stage experiences determining a drug's success—such as Phase 2b/3 trials, global regulatory strategies, FDA meetings, and commercialization preparation—were largely ceded to overseas partners. The recent move by some companies to take their pipelines through late-stage clinical trials into their own hands holds value in internalizing the experience and know-how that previously flowed overseas, thereby elevating Korea's drug development capabilities to the next level. Technology exports remain necessary and will continue to be a vital survival strategy. However, companies aiming to compete on the global stage should resolve to “see at least a few pipelines through to the end.” While not every biotech company can conduct late-stage clinical trials directly, simply accumulating one or two more companies with ‘full-cycle experience’ will undoubtedly elevate the standing of Korea's biotech sector. May technology transfer become not the final destination for survival, but the starting point toward completing the full cycle.