- LOGIN
- MemberShip
- 2026-07-22 08:53:52
- Company
- Enhertu expands treatment scope into HER2 solid tumors
- by Son, Hyung Min May 11, 2026 09:18am
- The HER2-targeted antibody-drug conjugate (ADC) ‘Enhertu’ is accelerating its expansion into the solid tumor market by broadening its indications in Korea.As Enhertu expands its indications to include first-line breast cancer and second-line gastric cancer treatment, there is growing speculation that treatment strategies for HER2-positive solid tumors may shift toward ADCs.ADC anticancer drug ‘Enhertu’According to industry sources on the 9th, the Ministry of Food and Drug Safety recently approved the expansion of Enhertu’s (trastuzumab deruxtecan) indications to include first-line therapy for HER2-positive metastatic breast cancer and second-line therapy for HER2-positive metastatic gastric cancer.This approval allows Enhertu to be used in combination with pertuzumab as first-line therapy for patients with unresectable or metastatic HER2-positive breast cancer, as well as for patients with HER2-positive gastric or gastroesophageal junction adenocarcinoma whose disease has progressed following trastuzumab-based therapy.Enhertu was previously approved as a second-line treatment for HER2-positive metastatic breast cancer and a third-line treatment for HER2-positive metastatic breast cancer.Enhertu is a next-generation ADC that combines a monoclonal antibody with the same structure as trastuzumab, which binds to specific target receptors overexpressed on the surface of cancer cells, with a highly potent topoisomerase I inhibitor payload via a tumor-selective cleavage linker.An ADC is a novel anticancer drug created by linking an antibody that binds to a specific target antigen on the surface of cancer cells to a drug (payload) with cytotoxic activity via a linker. This therapy has the advantage of enhancing treatment efficacy while minimizing side effects by leveraging the antibody’s target selectivity and the drug’s cytotoxic activity to ensure the drug acts selectively on cancer cells.For over a decade, the THP regimen, which is a combination of taxane chemotherapy, Herceptin (trastuzumab), and Perjeta (pertuzumab), has remained the standard first-line treatment for HER2-positive metastatic breast cancer. However, limitations have been noted, as a significant number of patients experience disease progression within two years, and some are unable to proceed to subsequent treatments.In the DESTINY-Breast09 study, which served as the basis for this approval, the Enhertu and Perjeta combination reduced the risk of disease progression or death by 44% compared to the existing standard THP regimen. The median progression-free survival (PFS) was 40.7 months, an extension of more than one year compared to 26.9 months in the THP group.In terms of response rates, the objective response rate (ORR) in the Enhertu combination group was 85.1%, higher than the 78.6% in the THP group, and the complete response (CR) rate was 15.1%, exceeding the 8.5% in the control group.At the European Society for Medical Oncology Asia Congress (ESMO Asia 2025) held last year, analysis results for the Asian patient population of the trial were also released. An analysis of 346 Asian patients, including those from South Korea, showed that the median PFS in the Enhertu plus Perjeta combination group was 40.7 months, reducing the risk of progression by 45% compared to the THP group’s 24.7 months.Potential shift in gastric cancer treatment strategiesEnhertu’s expansion is not limited to breast cancer. It is also showing potential to improve survival in HER2-positive gastric cancer, an area with long-standing unmet needs, which is raising expectations for a shift toward ADC-based treatment strategies.HER2-positive gastric cancer is considered a prime area of unmet medical need. While the 5-year survival rate for early-stage gastric cancer exceeds 90%, it drops sharply in the metastatic stage. According to national cancer registry statistics, the 5-year relative survival rate for patients with metastatic gastric cancer is only around 6–7%.Nevertheless, the treatment landscape for HER2-positive gastric cancer has remained largely unchanged for a long time since Herceptin plus chemotherapy became the standard first-line treatment in 2010. Although various HER2-targeted therapies have been developed since then, they have failed to demonstrate clinical outcomes in gastric cancer as clear as those seen in breast cancer.In fact, treatment strategies based on Perjeta, Kadcyla (trastuzumab emtansine), and lapatinib have all failed to achieve significant improvements in survival in gastric cancer clinical trials. As a result, HER2-positive gastric cancer has been considered a tumor type with limited responsiveness to HER2-targeted therapy.Amid this context, Enhertu demonstrated an improvement in overall survival (OS) in the second-line treatment of HER2-positive metastatic gastric cancer through the DESTINY-Gastric04 study. In the trial, Enhertu reduced the risk of death by 30% compared to the standard combination of Cyramza (ramucirumab) and paclitaxel, and the median OS was 14.7 months, an improvement over the 11.4 months observed in the control group.Progression-free survival (PFS) was 6.7 months versus 5.6 months, and ORR was 44.3% versus 29.1%, respectively.

- Policy
- Review initiated on introducing 'indication-based drug pricing'
- by Jung, Heung-Jun May 11, 2026 09:18am
- The National Health Insurance Service (NHIS) is proceeding with a fiscal impact review for the introduction of an indication-based pricing system. The final decision on whether to implement the system is expected to be determined by the results of this research.In March, the Health Insurance Policy Review Committee (HIPRC) decided to review the feasibility and effectiveness of indication-based pricing. Consequently, the NHIS has initiated follow-up measures by commissioning a research project.According to the NHIS on the 8th, a public bid has been announced for a research project titled “Analysis of Indication-Based Drug Price Evaluation Status and Feasibility Review Study,” which is to be completed by the end of this year. The research is scheduled to run for 5 months following contract signing, with a planned completion date between November and December.The NHIS plans to finalize the research project related to the of indication-based pricing system by November or December. There have been consistent criticisms that the current single-price operation of the national health insurance system has limitations in adequately reflecting the value of innovative new drugs for severe and rare diseases.Through this study, the NHIS will analyze the benefits and effects of improved accessibility under an IBP system, as well as the limitations and costs associated with allocating the health insurance budget. The research findings will serve as a reference for future policy decisions.The contents of the research consists of ▲Literature review of previous domestic and international studies related to IBP ▲Current status of IBP operations in major overseas countries ▲Objective measurement and analysis of patient accessibility benefits and fiscal impacts for multi-indication drugs ▲Opinion surveys and in-depth interviews with the public, academia, the pharmaceutical industry, and the government ▲Comprehensive policy recommendations for system improvement.Specifically, the NHIS plans to evaluate fiscal impacts through drug price simulations for each indication. The study will also cover the types of drugs to be included in the system, reimbursement evaluation, drug price calculation methods, and mechanisms for post-reimbursement expenditure management.Furthermore, the NHIS will examine the overall aspects of the indication-based pricing system, including its pros and cons, operational points of contention, and policy considerations.The NHIS anticipates that "We will derive mid-to-long-term development directions for the 'expansion of scope of use' system based on the categorization of reimbursement expansion for multi-indication drugs."The research results are planned for use in establishing a mid- to long-term roadmap for creating an innovative new drug ecosystem and in informing government policy decisions.The research schedule includes an interim report meeting in September~October and a final report meeting in October~November. The research is scheduled for completion at the end of the year.
- Opinion
- [Desk’s View] Are Korean modified drugs really beneficial?
- by Lee, Tak-Sun May 11, 2026 09:18am
- Generics with modified formulations are currently the hottest trend in product development among domestic pharmaceutical companies. In particular, products that convert tablets into orally disintegrating tablets (ODTs) to improve ease of administration are steadily emerging.ODTs, which dissolve in the mouth, are undoubtedly convenient for elderly patients, children, and those with swallowing difficulties. For these patients, there is no reason not to welcome the product development efforts of domestic pharmaceutical companies.However, whether it is really necessary for multiple companies to simultaneously develop orally disintegrating tablets containing the same active ingredient remains well in question, given the relatively small target patient population.For example, in the case of pitavastatin ODTs used to treat hyperlipidemia, not only the originator but also numerous generic companies have entered development competition, even though the 2mg tablet market is already saturated with 42 products.Behind the competition in ODT development lies pricing. In Korea, drug prices are calculated based on the criteria of identical active ingredients, identical dosage forms, and identical strengths. Under the stepwise pricing system, if there are more than 20 identical formulations meeting these conditions on the market, the price of the next product to be listed is set 15% lower than the previous lowest price.Since there are already 42 pitavastatin 2mg tablets, prices are inevitably set lower than the current minimum of KRW 462. However, ODT formulations, being classified as different formulations, can receive the highest price of KRW 561 for that ingredient.However, one must consider whether charging the highest price with a different formulation can truly satisfy a company’s profit needs when the patient population itself is small. Moreover, if multiple pharmaceutical companies compete, the market share will inevitably shrink.Naturally, to meet their profit targets with orally disintegrating tablets, pharmaceutical companies will target general populations as well, not just those who have difficulty swallowing tablets. As a result, a significant number of general patients may end up being prescribed ODTs.If a patient taking two or more medications has different dosage forms for each, this may increase inconvenience, as the patient must swallow one pill with water and dissolve another to take it, creating additional hassle.This means that a drug designed for the convenience of a specific patient group may actually cause inconvenience for the general patient population. This is because the choice of medication rests solely with the medical staff.The development of pharmaceuticals with improved convenience is on the rise. Although the market for combination drugs, which combine multiple medications into a single formulation, is already saturated, new combinations continue to emerge. Incrementally modified combination drugs are also granted pricing premiums. If ODTs prove commercially successful, development will likely evolve further.Pharmacy shelves are already filled with such products, and pharmacists have identified Korean modified drugs as a growing inventory burden. Given that hundreds of millions of won have been invested in developing these drugs, this cannot help but be a waste of social resources.While the emergence of more convenient medications is a positive development for patients, too many products are being released in Korea relative to the size of the market. In this highly competitive environment, it is unclear whether formulation changes and combination products truly contribute to the sustainability of the national health insurance system.If drug pricing is driving this competition in product development, the government may need to reassess whether the current pricing system truly aligns with patient needs.

- Company
- New drug competitiveness, patient-focused structure is the key
- by Hwang, byoung woo May 11, 2026 09:18am
- As the Korea Drug Development Fund (KDDF) enters its second phase, opinions have emerged that a structural shift, prioritizing higher success rates over simple pipeline expansion, is necessary for future tasks.During the investment review process, voices called for evaluating project quality based on patient population definition, clinical positioning, and biomarker strategies, rather than viewing innovation and commercialization potential as a completely distinguished one.On the 8th, the KDDF held the '2026 Investment Review Committee Workshop' and conducted a panel discussion under the title 'Investment Review that Produces Results: Directions and Choices for National Drug Development Projects."The Korea Drug Development Fund (KDDF) held the '2026 Investment Review Committee Workshop' on the 8th.The panel discussion was moderated by Koh Dae-kyung, Senior Manager at KDDF, with participation from: ▲Professor Sung Hoon Kim of Yonsei University ▲Junghee Lim, Vice President of InterVest ▲Professor Jaeho Cheong of Yonsei Cancer Hospital ▲Taegon Baik, CEO of Arum Therapeutics.Korean drug development, highlighting challenges in clinical trials and capital beyond pipeline countThe discussion began with an evaluation of Korea's competitiveness in drug development. Despite a rapid increase in domestic pipelines, participants discussed why global drug outcomes remain limited and identified the gap between R&D capabilities and actual performance.Panelists agreed that while the quantitative growth of the domestic ecosystem is evident, it must be supplemented by Phase 3 capital, commercialization judgment, and patient-centric development strategies to translate into global success.First, Professor Sung Hoon Kim noted that the domestic ecosystem has grown to an incomparable scale, but cautioned that quantitative expansion does not automatically yield global results. Professor Kim said, "With thousands of pipelines in motion, even if most fail statistically, I believe a global drug will emerge soon," and assessed, "However, for this to be structurally possible, we ultimately need funds capable of supporting Phase 3 trials."InterVest Vice President Junghee Lim noted that the increase in pipelines should be viewed alongside its underlying structure. Lim explained that many projects are likely in early clinical stages, often linked to the rise of university-based venture startups. While acknowledging the advantage of having the most knowledgeable technical experts lead development, Lim questioned whether rigorous eligibility judgments from a commercialization perspective were adequately applied.(from left Koh Dae-kyung, Senior Manager at KDDF, Professor Jaeho Cheong of Yonsei Cancer Hospital , Professor Sung Hoon Kim of Yonsei University From a clinical perspective, 'patient-centric' was presented as a key keyword. Professor Jaeho Cheong suggested that, since the endpoint of drug development is the patient, the approach should shift away from a 'substance-centric' focus toward defining which patient groups, which biomarkers, and which clinical benefits will be achieved. Professor Cheong stated, "We are playing a game of probability regarding patient reachability," and added, "We must move toward a patient-centric, not substance-centric, approach."Professor Cheong added that competitiveness should be viewed based on clinical benefit rather than the number of candidates. This means that a focus on which patient segments to target and what clinical positioning to take must come first to increase the value of thousands of drugs.Role of investment review…balancing innovation and failure ProbabilityFollowing the evaluation of competitiveness, the discussion moved to the roles of the Fund and the Investment Review Committee. Senior Manager Koh Dae-kyung asked which criteria (among global competitiveness, innovation, performance potential, or commercialization potential) should be the primary focus.The discussion focused on the balance between 'selecting projects with high success probability' and 'early screening of projects with high failure probability.' As a project funded by public capital, it cannot focus solely on short-term results, yet it is also inappropriate to delay the verification of development feasibility in the name of innovation.Professor Chung proposed 'Translational Probability,' the likelihood of reaching the patient, as the core criterion for investment review. He explained that the role of public support is not just picking winners but also screening out projects with high failure probability early on."The endpoint of the risky journey of drug development is reaching the patient," Professor Chung explained. "Creating criteria that can quantify, index, and objectify this will be the role of the next generation of the Investment Review Committee." However, considering that the KDDF is at a stage where it must produce results in its remaining period, he acknowledged that the weight of competitiveness and commercialization potential might increase.(from left) Junghee Lim, Vice President of InterVest, Taegon Baik, CEO of Arum TherapeuticsFrom an investor's perspective, competitiveness and success probability were emphasized more heavily. Vice President Lim explained that the committee is composed of experts from various fields to view a single project from multiple perspectives. Lim stated that if a 'Best-in-class' drug is targeted, the proposal must include head-to-head comparisons with competitors, a mechanism of action (MoA) demonstrating competitiveness, evidence-based experimental models, and clinical trial designs.Regarding novel targets, Lim also distinguished between research and development. While a novel target may be a research topic to explore over a long period for academia, a company must produce results within limited resources and time.Future Projects, re-designing support systems to increase clinical success ratesIn the latter half of the discussion, participants deliberated on the direction of support after the current national drug development project and the structure of subsequent projects.Panelists argued that future projects should be designed to increase the clinical success rate, moving beyond simple research funding. Key tasks identified included investment linkage, clinical site matching, platform technology support, and strategies for securing Human Proof of Concept (PoC).Regarding this, CEO Taegon Baik suggested different support methods for discovery and clinical stages. Since the discovery stage is high-risk, it should be linked to corporate strategic investment and joint research. At the same time, national budgets should be more concentrated on clinical programs with high success potential.Professor Kim also proposed a separate track for platform technology support. Unlike individual pipelines, platform technologies have independent value but can appear ambiguous from a private investment perspective, necessitating a dedicated track for public support.Park Yeong min, Director of the Korea Drug Development FundDuring the Q&A session, issues such as securing financial resources for subsequent projects, operating expert advisory groups, public-led risk-sharing systems, and the need for a legal foundation were raised. Attendees suggested that since government budgets alone have limits, there should be linkages with long-term investment funds like the National Pension Service and expanded participation from private capital.Park Yeong min, Director of the Korea Drug Development Fund, stated that securing innovation, balancing evaluation and management, recycling budgets from halted projects, and ensuring the continuity of expert organizations are all tasks to be addressed in subsequent projects."We need to consider how to secure innovation and what evaluation criteria to maintain since public funds are involved," Director Park said. "From a VC's perspective, early exits may be important, while from a developer's perspective, there is a need for support until the end to develop a 'First-in-class' drug."Park added, "There are projects that are discontinued during stage or final evaluations, and we need wisdom on how to reuse those halted budgets. We must prepare in a way that preserves the strengths identified so far while minimizing the weaknesses."
- Company
- Rybrevant reimb delayed in Korea…treatment gap persists
- by Son, Hyung Min May 11, 2026 09:18am
- NSCLC drug ‘Rybrevant’The gap in treatment access continues as discussions on health insurance reimbursement for ‘Rybrevant,’ a treatment for non-small cell lung cancer (NSCLC) with EGFR exon 20 insertion mutations, have been postponed again.This mutation is known for its low response rate to conventional EGFR-targeted therapies and for the limited treatment options available. With virtually no alternatives to Rybrevant currently available, reimbursement delays continue to place a financial burden on patients.According to industry sources on the 11th, the Drug Reimbursement Evaluation Committee of the Health Insurance Review and Assessment Service (HIRA) recently issued a redeliberation decision regarding the adequacy of reimbursement for Janssen’s NSCLC treatment, Rybrevant (amivantamab).The DREC evaluated its use as monotherapy for patients with locally advanced or metastatic NSCLC harboring EGFR exon 20 insertion mutations whose disease has progressed during or after platinum-based chemotherapy.Although Rybrevant received regulatory approval in Korea in December 2022, it has yet to obtain reimbursement coverage.In addition, the Cancer Drug Deliberation Committee meetings held in September last year and January this year failed to establish reimbursement criteria for several regimens, including ▲ first-line carboplatin + pemetrexed combination therapy for EGFR exon 20 insertion patients; ▲ first-line lazertinib combination therapy for EGFR exon 19 deletion or L858R mutation patients; and ▲ carboplatin + pemetrexed combination therapy following EGFR TKI treatment.Setbacks in development of exon 20 insertion targeted therapies… Rybrevant remains the only optionEGFR exon 20 insertion mutations are structurally complex and heterogeneous compared to exon 19 deletions or L858R mutations, making drug development particularly challenging.In fact, cases of failed drug development have continued in the global market as well.Takeda’s oral targeted therapy Exkivity (mobocertinib) initially received conditional approval based on an objective response rate (ORR) of 28% in early trials, but was withdrawn after failing to demonstrate improvement in progression-free survival (PFS) in the confirmatory Phase III EXCLAIM-2 study.Development of poziotinib was also halted due to efficacy falling short of expectations and toxicity issues.Amid these challenges, Rybrevant has effectively become the only approved treatment option in this setting.Rybrevant is a bispecific antibody targeting both EGFR and MET. It is designed to inhibit not only EGFR mutations but also MET-driven resistance pathways.In clinical practice, MET-based resistance mechanisms are observed in approximately 10–15% of all patients. This patient group has a relatively poor prognosis, and Rybrevant is therefore considered to offer meaningful potential in terms of long-term survival.Rybrevant shows efficacy as first-line combination therapy… patient burden remainsWhile reimbursement discussions for Rybrevant are delayed, clinical practice is increasingly focusing on the value of first-line combination therapy rather than monotherapy.In the Phase III PAPILLON study, Rybrevant in combination with pemetrexed and carboplatin improved both PFS and ORR compared to chemotherapy. The trial included 308 previously untreated patients with locally advanced or metastatic NSCLC harboring EGFR exon 20 mutations.In this study, median PFS was approximately 11.4 months in the combination group versus 6.7 months in the chemotherapy group.However, since current reimbursement discussions are focused on its use as second-line monotherapy, some observers note a gap between the pace of accumulating clinical evidence and policy application.The continued lack of reimbursement imposes a significant financial burden on patients. Annual treatment costs for Rybrevant without reimbursement are estimated at around KRW 150 million.In particular, since the drug is administered once a week during the first four weeks, the financial burden is concentrated in the early treatment phase. After this initial phase, the dosing schedule switches to every two weeks. However, it is reported that the pharmaceutical company is currently operating a partial cost-support program for the initial treatment phase, thereby reducing the actual financial burden on patients. Under this program, a portion of the drug cost is covered during the first four weeks, and a certain percentage of the cost is also covered during the subsequent maintenance therapy phase.Industry observers suggest that Janssen is placing greater strategic emphasis on its use as a combination therapy, which is gaining traction in clinical practice, rather than focusing solely on its monotherapy reimbursement.Rybrevant is currently being developed in combination with Leclaza to expand into first-line treatment of EGFR-mutant NSCLC, while also broadening the potential for its use in the early treatment stages for Exon 20 insertion mutations.However, while treatment strategies in clinical settings are shifting toward first-line combination therapy, reimbursement discussions remain stuck at second-line monotherapy, highlighting an ongoing disconnect between policy and clinical practice.
- Policy
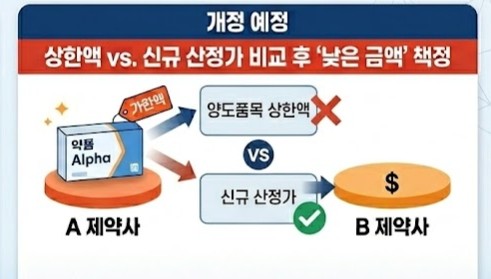
- Inheritance of drug price ceiling to be blocked in transfers
- by Jung, Heung-Jun May 08, 2026 01:15pm
- Changes are expected to the current rules that allow drug price ceilings to be inherited during product transfers. The system is likely to revert to past regulations that apply the lower of the new calculated price and the price at the time of transfer.As a result, acquiring high-priced generics to avoid the impact of drug pricing reform will no longer be viable.According to industry sources on the 6th, a working-level consultative body comprising the Ministry of Health and Welfare and industry representatives recently discussed changes to the transfer regulations that were not brought before the Health Insurance Policy Deliberation Committee in March.The rules governing price ceilings in transfers have changed multiple times. Until 2020, prices were set based on the lower of the final ceiling price of the transferred product and the newly calculated price.However, following the stepwise pricing reform, industry backlash arose as transfers were treated as new listings, leading to significant price drops.In response, the government allowed inheritance of the existing ceiling price starting during transfers in 2021.Now, the government plans to revert to the previous rule, applying the lower of the existing ceiling and the new calculated price.Even if a high-priced generic is acquired, the newly calculated price at the moment of transfer is applied, effectively enforcing a lowest price rule.Pharmaceutical companies that had considered acquiring high-priced generics to avoid pricing reform will lose this option.With the reform, the generic pricing rate has been reduced to 45%, and the reduction rate for products failing to meet the baseline requirements has been strengthened from 85% to 85%. Combined with multi-listing management rules, prices could fall to as low as 30.6% within one year if bioequivalence studies are not conducted.Previously, companies might have chosen to acquire products with higher maintained prices.However, if prices are reset based on the calculation price at the time of transfer, transfers that take existing drug prices into account become meaningless.
- Company
- Hunter syndrome drug Hunterase ICV approved in Peru
- by Lee, Seok-Jun May 08, 2026 01:15pm
- GC Biopharma announced on the 6th that its intracerebroventricular (ICV) Hunter syndrome treatment, ‘Hunterase ICV’, has received marketing approval from Peru’s regulatory authority, General Directorate of Medicines, Supplies and Drugs (DIGEMID).This marks the third overseas approval following Japan and Russia. The company plans to expand into additional countries using its Latin America entry as a foothold.Hunter syndrome is a rare inherited disease caused by a deficiency of the IDS (Iduronate-2-sulfatase) enzyme, leading to the accumulation of glycosaminoglycans (GAGs). It presents with skeletal abnormalities, joint deformities, respiratory and cardiac dysfunction, and cognitive impairment. It is known to occur in approximately 1 in every 100,000 to 150,000 male infants.About two-thirds of patients develop severe forms involving central nervous system damage. As the disease progresses, cognitive decline and behavioral abnormalities emerge, affecting patients’ quality of life and prognosis.Hunterase ICV is administered directly into the brain ventricles once a month. In Japanese clinical trials, it significantly reduced heparan sulfate, a key factor in CNS damage.The company also reported stabilization or improvement in cognitive and developmental functions, with sustained efficacy confirmed in long-term follow-up.Jae-woo Lee, head of R&D at GC Biopharma, said, “Based on long-term clinical data, we will focus on addressing the unmet medical needs of patients with severe Hunter syndrome.”
- Company
- SK bioscience's successful M&A
- by Chon, Seung-Hyun May 08, 2026 01:15pm
- The sales share of SK bioscience’s German contract development and manufacturing organization (CDMO) subsidiary has reached nearly 80%. By deploying funds secured during the COVID-19 pandemic into mergers and acquisitions (M&A), the company has strengthened its financial position and improved its defense against market pressures.According to SK bioscience, on the 7th, IDT Biologika’s first-quarter sales reached KRW 128.3 billion, an 8.5% increase from the same period last year.IDT Biologika is a German biotech company acquired by SK bioscience in 2024. SK bioscience purchased a 60% stake in IDT Biologika, previously held by the German biopharmaceutical firm Klocke Gruppe, through a wholly owned German subsidiary. The total acquisition price for IDT Biologika amounted to KRW 370.0 billion. SK bioscience decided to allocate paid-in capital to a third party, issuing 1,519,543 new shares worth KRW 75.7 billion to the Klocke Gruppe. Consequently, the total cash invested by SK bioscience in the acquisition of IDT Biologika is KRW 294.3 billion.Sales of SK bioscience and IDT Biologika (unit: KRW 100 million, source: SK bioscience) BLUE: SK bioscience, GREEN: IDT BiologikaFounded in 1921, IDT Biologika is a major biotech company operating CDMO businesses in Germany and the United States. It has a track record recognized by more than 10 core drug regulatory agencies, including those in the U.S. and Europe. The company develops processes and analytical methods while producing drug substances (DS) and drug products (DP) across the entire vaccine and biotech spectrum from clinical to commercial stages, employing approximately 1,800 people.IDT Biologika’s performance began to be reflected in SK bioscience’s consolidated financial statements starting from the fourth quarter of 2024. Revenue from IDT Biologika of KRW 111.2 billion was recognized in Q4 2024, and last year it recorded revenue of KRW 465.7 billion.With the inclusion of IDT Biologika’s results, SK bioscience’s revenue surged. In the first quarter of this year, SK bioscience’s revenue expanded more than sevenfold compared to the first quarter of 2024.SK bioscience faced significant fluctuations in performance throughout the COVID-19 pandemic.In 2020, SK bioscience signed a contract manufacturing agreement with AstraZeneca to produce and supply COVID-19 vaccine drug substances and finished products. In the same year, SK bioscience signed a tripartite agreement with the Ministry of Health and Welfare and Novavax to supply the COVID-19 vaccine 'NVX-CoV2373,' beginning full-scale contract manufacturing and supply in 2021.SK bioscience’s sales in the first quarter of 2020 was only KRW 22.7 billion. Then, it jumped nearly fivefold over the year to KRW 112.7 billion in the first quarter of 2021, and then rose to KRW 450.9 billion in the fourth quarter of 2021.However, as the contract manufacturing performance for COVID-19 vaccines dissipated, sales dropped. In the first quarter of 2023, SK bioscience’s sales amounted to KRW 20.6 billion, a 95.4% reduction compared to the fourth quarter of 2021.While sales fell sharply after the end of the pandemic, the M&A strategy successfully minimized the sales gap. In the first quarter of this year, IDT Biologika accounted for 76.1% of SK bioscience’s total sales. IDT Biologika’s sales were more than three times higher than SK bioscience’s separate sales of KRW 40.3 billion.From the fourth quarter of 2024 to the first quarter of this year, IDT Biologika’s cumulative sales reached KRW 705.2 billion, accounting for 72.2% of the parent company's sales.
- Policy
- Innovative pharmas need permanent pricing incentives
- by Lee, Jeong-Hwan May 08, 2026 01:15pm
- Rep. In-soon Nam of the Democratic Party of Korea has drawn attention by stating that permanent pricing incentives for innovative pharmaceutical companies should be institutionalized in order to advance Korea into a pharmaceutical and biotech powerhouse and create blockbuster new drugs.Rep. Nam criticized the Ministry of Health and Welfare, noting that while the ministry recently formalized a differential drug pricing system between innovative and non-innovative pharmaceutical companies through a resolution by the Health Insurance Policy Deliberation Committee, the inclusion of preferential pricing for innovative pharmaceutical companies only as a temporary provision left some gaps.Regarding legislation to introduce limited international nonproprietary name (INN) prescribing, Rep. Nam also expressed the view that it would be reasonable to either remove penalty provisions for doctors who fail to comply with mandatory INN prescribing regulations or to downgrade and relax these penalties to administrative fines.Her point is that policy and legislation should be pursued with a focus on ensuring that the public can access frequently out-of-stock and essential medicines without barriers, setting aside conflicts between specific professions such as doctors and pharmacists.Having officially announced her candidacy for Deputy Speaker of the second half of the 22nd National Assembly, Rep. Nam In-soon promised that, if elected, she would strive to create a national environment that advances the pharmaceutical and biotech industry and enhances public access to essential medicines.On the 7th, Rep. Nam held a meeting with the press corps and stated, “As the Jae-myung Lee administration has adopted a national agenda to transform Korea into a leader in digital health and the pharma-biotech industry, I will consider various ways to support this goal as Deputy Speaker.”“Pharma-biotech lost priority under Yoon administration… will restore momentum”Rep Nam assessed that while fostering the pharma-biotech industry has always been a priority across administrations, interest declined under the former Yoon, weakening growth momentum.In contrast, she noted that the Lee administration has set a goal of becoming one of the world’s top 5 pharma powerhouses and developing domestic blockbuster drugs, raising expectations for policy reforms across related ministries, including the Ministry of Health and Welfare.She therefore argued for the need to permanently extend preferential drug pricing for innovative pharmaceutical companies with the goal of fostering blockbuster new drugs.Rep Nam stated, “I have consistently advocated for differential pricing between innovative and general pharmaceutical companies, and I welcome its inclusion in the recent reform. However, making the incentive temporary limits its impact on new drug R&D investment.”“The government’s goal is to improve the domestic pharmaceutical industry’s structure using the funds secured through drug price cuts. If we want innovative new drugs and blockbusters to emerge in Korea, preferential treatment for innovative pharmaceutical companies cannot be temporary. Instead of offering temporary preferential treatment to innovative companies, we must remove the ‘temporary’ qualifier to allow for active support.”Rep Nam also noted that the Health and Welfare Minister Eun-Kyeong Jeong has shown a strong commitment to fostering domestic new drug development.She said, “The Lee administration has declared its intention to nurture blockbuster drug companies and has merged the Bio Committee and Bio-Health Innovation Committee. Previously, the Ministry of Health and Welfare did not appear to take a leading role, but after speaking with Minister Jeong Eun-kyung following the integration of the innovation committees, it appears the Ministry intends to take the initiative and carry out its duties. The atmosphere will change going forward.”He noted, “Currently, South Korea lags behind advanced countries in terms of pharmaceutical technology, new drug development, and medical AI, so we must catch up quickly. Since I enacted the Special Act on the Promotion of the Pharmaceutical Industry, preferential measures have been stalled for several years. If elected Deputy Speaker, I will prioritize this area in my legislative work.”“Let’s carry out limited INN prescribing focused on patients, without provisions punishing doctors”Regarding the limited INN prescribing bill currently pending in the National Assembly’s Health and Welfare Committee, Rep Nam expressed the view that even if provisions punishing doctors who violate the mandatory generic prescribing requirement are removed, a legislative environment must be created that ensures patients can access essential medicines without inconvenience.She emphasized that, since limited INN prescribing is a key policy agenda item of the current administration, efforts should focus on addressing resistance from the medical community and ensuring smooth institutional adoption.The current bill mandates INN prescribing for selected drugs designated by the Ministry of Health and Welfare following a government committee review, and stipulates penalties of up to one year in prison or a fine of up to KRW 10 million for violations.Rep Nam clearly stated that these penalty provisions should either be removed entirely or downgraded to administrative fines.She also noted the need to carefully consider concerns raised by the Korean Medical Association regarding the scope of bioequivalence testing and trust in substitution and INN prescribing.Rep Nam said, “The KMA is opposing the measure, raising concerns about the broad scope of bioequivalence testing, and their arguments should be taken seriously. We need further discussion on how to incorporate these concerns into policy. For now, it seems there is a lack of objective data or research on generic substitution and INN prescribing, so we need to accumulate more data.”She added, “Gradual implementation of INN prescribing for out-of-stock and essential medicines could serve as a middle ground between physicians and pharmacists. “Public awareness of generic substitution and INN prescribing has significantly increased during the COVID-19 pandemic, as more people have experienced receiving alternative drugs with the same active ingredient.”Nam emphasized, “We should activate systems such as post-notification of substitution and build more research data to support future legislative discussions. But imposing criminal penalties for INN prescribing violations is inappropriate. A more reasonable approach would be to remove penalties or reduce them to administrative fines, prioritize patient-centered legislation, and consider enforcement measures in stages.”“Warehouse pharmacy regulations need to prevent misuse”The recent amendment to the Pharmaceutical Affairs Act, which strengthens regulations on warehouse-style pharmacy advertising, is also one of Nam’s achievements.She explained that the ultimate goal of the regulation is to prevent excessive consumption and misuse of medicines that could lead to adverse effects.The provision in the Pharmaceutical Affairs Act prohibits expressions that may encourage misuse and delegates authority to the Ministry of Health and Welfare to specify concrete and unique expressions that cannot be used in pharmacy names or promotional slogans through subordinate regulations, prioritizing public safety above all else.Rep Nam said, “We also brought forward the implementation date from six months to three months after the government’s promulgation, and added a supplementary provision allowing public health centers to provide administrative guidance to pharmacists opening new pharmacies before the law takes effect. I was the one who codified the provision for pharmacists to provide medication guidance within the Pharmaceutical Affairs Act. At first, pharmacists weren’t very fond of it, but today, medication guidance has become a crucial system and phrase for ensuring public medication safety.”She continued, “When I look at warehouse-style pharmacies, I'm sorry whether pharmacists can actually provide proper medication guidance to patients. We must prevent the public from perceiving medications as mere commodities. Even after my election as Deputy Speaker, I will engage in policy monitoring to curb the misuse of medications by the public by regulating warehouse-style pharmacies.”

- Company
- 'Super antibiotic' Fetroja enters the prescription lists of gen hospitals
- by Eo, Yun-Ho May 08, 2026 01:15pm
- The 'super antibiotic' Fetroja is entering the prescription lists of major general hospitals.According to industry sources, Fetroja (cefiderocol tosylate sulfate), Jeil Pharmaceutical’s treatment for multidrug-resistant Gram-negative bacterial infections, has passed the drug committees (DC) of tertiary general hospitals, including Samsung Medical Center and Seoul National University Hospital.Following its inclusion on the insurance reimbursement list last February, the drug is gradually expanding its prescribing area.Fetroja was approved in South Korea in February 2025 for the treatment of ▲complicated urinary tract infections, including pyelonephritis ▲hospital-acquired pneumonia, including ventilator-associated pneumonia.Developed by Shionogi, Fetroja is the world's first siderophore cephalosporin antibiotic. To overcome reduced efficacy caused by bacterial resistance mechanisms, it uses a unique mechanism of action: it binds to ferric iron. It is actively transported into the bacterial cell through the pathogen's own iron porin channels.Jeil Pharmaceutical secured the rights to develop and commercialize Fetroja in South Korea after signing an exclusive domestic supply agreement with Ping An-Shionogi in July 2022.Fetroja has demonstrated in vitro activity against various antimicrobial-resistant (AMR) pathogens, including Carbapenem-resistant Enterobacterales (CRE), Carbapenem-resistant Acinetobacter baumannii (CRAB), and Metallo-beta-lactamase (MBL)-producing Carbapenem-resistant Pseudomonas aeruginosa (CRPA).Meanwhile, Shionogi has a long history and expertise in developing treatments for infectious diseases. The company continues to conduct research and development, particularly in antibiotics, antivirals, and central nervous system (CNS) treatments, and maintains subsidiaries in Japan, the U.S., Europe, and China.Ping An-Shionogi was established as a joint venture between Japan's Shionogi and China's Ping An in 2020. Ping An-Shionogi obtained Asian rights to Fetroja. In December 2024, Shionogi acquired all of Ping An's shares, and Ping An was subsequently incorporated as a wholly owned subsidiary of Shionogi.