- LOGIN
- MemberShip
- 2026-05-04 22:01:28
- Company
- Autotelic Bio signs licensing agreement with Chinoin
- by Nho, Byung Chul Jun 12, 2024 05:45am
- (From the left) Fernando Torres Navarrete, BD Director at Chinoin Productos Farmaceuticos in Mexico , Tae-Hun Kim, CEO of Autotelic Bio Autotelic Bio announced on the 7th that it has signed an exclusive license and supply agreement with Chinoin Productos Farmaceuticos in Mexico to distribute ATB-101, a novel combination drug for the treatment of hypertension and diabetes. ATB-101 is a fixed-dose combination product comprising of the hypertension drug ‘olmesartan’ and the oral diabetes drug ‘dapagliflozin.’ It is the world's first combination drug that treats both hypertension and Type 2 diabetes, making it easier for patients with both chronic diseases to take their medication. The company has received Phase III IND approval from the Ministry of Food and Drug Safety for ATB-101 and is currently conducting Phase III clinical trials in patients with essential hypertension and type 2 diabetes at more than 35 major hospitals in Korea, including Seoul National University Bundang Hospital. Autotelic Bio is planning to launch the product globally after proving safety and efficacy through the ongoing Phase 3 clinical trial. For this, the company has registered composition patents in the U.S., Japan, Russia, Mexico, and Brazil, in addition to Korea, and is expanding its patent to other countries such as China. The exclusive license and supply agreement it signed in Mexico is to supply more than 30 million ATB-101 tablets over 5 years. The agreement demonstrates the company's potential to expand into neighboring Latin American countries through Chinosa and is expected to serve as a bridgehead for future global expansion. In addition to the upfront payment, Autotelic Bio will receive additional payments based on development, licensing, and commercialization milestones. Mexico's hypertension and diabetes market is the second largest in Latin America after Brazil and has significant potential. Autotelic Bio plans to grow sales in the Mexican market and reinvest the proceeds in the continued development of innovative new medicines. Chinoin Productos Farmaceuticos is a Mexican pharmaceutical company founded in 1924. It owns a pharmaceutical R&D, manufacturing, licensing, sales, and marketing division, and is regarded to have excellent sales and marketing capabilities, ranking among the top 10 pharmaceutical sellers in Mexico. Autotelic Bio recently completed a pre-IND meeting with the U.S. Food and Drug Administration (FDA) to advance ATB-101 into the United States. During the meeting, the company received a positive response from the FDA regarding the ongoing Phase III clinical trial, and that an additional local Phase I clinical trial will be sufficient for approval in the U.S. based on the results of the domestic Phase III trial. Based on the results of the pre-IND meeting, the company will be meeting with more than 10 pharmaceutical companies at Bio USA to discuss global rights agreements for ATB-101, including in the US. This year, Autotelic Bio has joined the Boston C&D Incubation Office, a blockbuster global expansion program organized by the Korea Health Industry Development Institute (KHIDI), to strengthen its network for ATB-101’s clinical development and licensing-out activities and create results in the U.S. market. In addition to the incrementally modified drug, Autotelic Bio is developing two other RNA-based anti-cancer drugs, ATB-320 (a dual-mechanism of action anti-cancer RNA drug that works on the TME and inhibits angiogenesis) and ATB-350 (a next-generation KRAS mutation-targeting anti-cancer RNA drug that targets specific tissue). Also, its ATB-610 (ALK5 inhibitor), an inhaler in the company’s anti-fibrosis drug pipeline, has been selected as a project of the National New Drug Development Project in Korea, adding momentum to the company’s development of new drugs.
- Company
- Failed reimb negotiations for Lorviqua, was it for the best?
- by Eo, Yun-Ho Jun 12, 2024 05:45am
- After a long wait, the result was still a ‘no go.’ A solution does exist, but hesitation seems to be holding them back. The insurance reimbursement expansion for the 3rd-generation ALK anticancer drug Lorviqua to first-line therapy has become unclear again. Negotiations between Pfizer Korea and the National Health Insurance Service on the drug price of the ALK-positive NSCLC treatment Lorviqua (lorlatinib), which passed the Health Insurance Review and Assessment Service’s Drug Reimbursement Evaluation Committee review in January and began in March, recently broke down. This is the first time negotiations have broken down for a drug seeking reimbursement expansion risk-sharing agreement (RSA) scheme. The reason for the breakdown is believed to be related to the ‘expenditure cap amount’ rather than 'drug price'. Lorviqua was granted pharmacoeconomic evaluation exemptions when it was first listed. Such PE exemption drugs are required to be reimbursed through the RSA Expenditure Cap Type scheme. As such, a new cap amount would have been derived to account for the increased usage due to the expanded reimbursement during negotiations, and it is likely that Pfizer was unable to accept it. Pfizer's unacceptance may be seen as greed on the pharmaceutical company’s part. However, negotiations are always conducted within a set framework. Pfizer did conduct a pharmacoeconomic evaluation as part of the reimbursement expansion process. So the solution would be to eliminate the disputed expenditure cap requirement and convert Lorviqua to a drug listed through the general process. The other ALK anticancer drugs available in the market - the 1st generation drug ‘Xalkori (crizotinib),’ 2nd generation drug ‘Alecensa (alectinib),’ ‘Zykadia (ceritinib)’ – were all listed through the general track. In fact, Pfizer offered to switch Lorviqua’s reimbursement path to general listing during negotiations, but the government turned it down as it was "unprecedented," and that hesitation led to the breakdown of negotiations. However, patients are left to suffer the damage. It's important to make things work. If necessary, there's no reason not to take the road less traveled. Moving RSA drugs to the general track will also ensure additional price transparency. Pfizer quickly announced plans to reapply for reimbursement expansions. A company spokesperson said: "We are disappointed that final negotiations were unsuccessful. We plan to reapply for first-line reimbursement as soon as possible to ensure patient access to the treatment and we will do our best to engage in effective discussions with the government and facilitate a forward-looking review.” As such, its progress upon reapplication will also be interesting to watch. This was the e first time RSA negotiations have failed. After the unprecedented event, starting the process all over again would be a lengthy process. Regardless of whether reimbursement is expanded or not, the government would need to be flexible in its administration to achieve a quick result. Lorviqua was specifically designed and developed by Pfizer to penetrate the blood-brain barrier (BBB). The drug’s high clinical value as a first-line treatment was recognized in the 5-year long-term follow-up results of the CROWN study that was presented at ASCO. Results showed that Lorviqua reduced the risk of disease progression or death by 81% compared to crizotinib, with 60% of patients surviving without disease progression at 5 years. The risk of brain metastasis progression was reduced in 94% of patients, with only 4 of 114 Lorviqua-treated patients without brain metastases developing brain metastases.

- Policy
- Price ceiling for 'atosiban' set to rise under agreements
- by Lee, Tak-Sun Jun 12, 2024 05:45am
- Hanlim Pharm’s The insurance drug price ceiling for atosiban injection, used to delay pre-term birth in pregnant adult women, will increase. Currently, three products of atosiban for injection are available. With the drug price ceiling adjustment, better supply is expected following negotiations related to expanding the volume of distribution. According to the industry on June 11, the drug price ceiling of three products containing atosiban acetate will rise. Three items include Ferring Korea’s 'Tractocile Inj,' Hanlim Pharm’s 'Trecsiban Inj,' and Dong Kook’s 'Asiban Inj.' These products are used to delay pre-term birth in pregnant adult women with ▲regular uterine contractions of at least 30 seconds duration at a rate of more than 4 per 30 minutes ▲a cervical dilation of 1 to 3 cm (0-3 for nulliparas) and effacement of greater than 50% ▲18 years and older ▲pregnancy 24-33 weeks, and ▲a normal fetal heart rate. It is an inhibitor of oxytocin and selectively acts on uterine muscle, potentially preventing uterine contractions. The drug is currently reimbursed when used as the second-line treatment following the use of a ritodrine-containing agent. It is not covered with reimbursement from week 3 and results in a burden of over KRW 500,000 per 1 cycle. Therefore, the medical community has been advocating for expanding reimbursement coverage for first-line treatment and making it reimbursable after week 3. Current adjustment negotiations have reached an agreement on an increased drug price ceiling based on the volume of contract manufacturing (imports). It appears that this measure is intended to stabilize the supply. The drug price ceiling for a 6.75 mg product will be increased from KRW 13,000-14,000 to KRW 15,000, and for a 37.5 mg product, it will be increased from KRW 43,000 to 47,000. As the government focuses on policies related to raising birth rates and supporting pregnant women, raising the drug price of pre-term birth treatment likely happened quickly. Starting this month, five products for morning sickness are also reimbursable.
- Company
- Boryung wins the Pomalyst patent challenge for the 2nd time
- by Kim, Jin-Gu Jun 11, 2024 03:25pm
- Boryung has once again challenged the Pomalyst patent and won. Boryung won the second attempt to avoid the substance patent of ‘Pomalyst (pomalidomide),‘ a treatment for multiple myeloma. Despite winning the infringement trial for the same patent in 2021, Boryung had not released generic versions. It seems that Boryung changed the method of generic development and rechallenged the patent. The analysis suggests that Boryung is on a countdown to releasing Pomalyst generic following the second-time success of the patent infringement challenge. It has been confirmed that Boryung has already applied for marketing approval for four doses of pomalidomide. According to pharmaceutical industry sources on June 10th, Boryung received the decision in its favor regarding the claims to confirm the scope of a right for the Pomalyst substance patent. Interestingly, Boryung has already won the previous challenge for the same patent. In July 2020, Boyung filed claims to confirm the scope of a right for the Pomalyst substance patent and won the claim in February. At that time, Kwang Dong Pharmaceutical also challenged the patent and won. However, Kwang Dong Pharmaceutical and Boryung have not released the generic version. Although the patent holder did not appeal the loss of the first trial, neither company released the generics. In three years, Boryung filed a claim with the Intellectual Property Trial and Appeal Board (IPTAB) again. The target for claiming a trial and the type of claim were the same as before. Regarding this, the pharmaceutical industry suggested that both companies attempted to develop Pomalyst generics using a method approved by the IPTAB but faced challenges. Boryung might have developed a generic using a different method. Developing a generic with a new method may have required a new trial decision to confirm whether it does not fall under the scope of the rights of the Pomalyst patent. Consequently, the company may have rechallenged the same patent and won. It has been confirmed that Boryung has already applied for marketing approval for its four doses of generics. The pharmaceutical industry anticipates that Boryung will secure a right to priority of sale for Pomalyst generic exclusively. Celgene’s Pomalyst was approved in 2014 for the treatment of multiple myeloma. According to a pharmaceutical market research firm IQVIA, Pomalyst’s sales reached KRW 22.8 billion last year, up 17% from KRW 19.5 billion in 2022.
- Product
- Selection dilemma rises in IBD mkt due to increased options
- by Moon, sung-ho Jun 11, 2024 05:48am
- With the recent surge in treatment options for inflammatory bowel disease (IBD), which is represented by ulcerative colitis and Crohn's disease, developing an appropriate treatment strategy for IBD is emerging as a rising topic in clinical practice. This is due to the recent health insurance reimbursement extensions granted for treatments by multinational pharmaceutical companies, which have lowered the burden on site. #Amid intensifying sales and marketing competition within the industry, the medical community is expected to revise its guidelines on selecting appropriate treatments. Until now, tumor necrosis factor (TNF) blockers Humira (adalimumab) and Remicade (infliximab) have dominated the field of IBD treatment in Korea. According to the pharmaceutical industry and medical community on the 8th, the IBD treatment market has been rapidly reshaping with the competitive entry of global pharmaceutical companies’ treatments this year. First, in the first half of this year, Lilly Korea received approval for its interleukin-23 (IL-23) inhibitor ‘Omvo (mirikizumab) from the Ministry of Food and Drug Safety. This added another IL inhibitor option to ‘Stelara (ustekinumab),’ the only anti-interleukin drug that had been available in the market until then. As a result, the anti-integrin agent ‘Kynteles (vedolizumab, Takeda),’ and anti-interleukin agent ‘Stelara,’ ‘Omvo,’ and the Janus kinase (JAK) inhibitors ‘Xeljanz (tofacitinib, Pfizer),’ ‘Rinvoq (upadacitinib, AbbVie),’ ‘Jyseleca (filgotinib, Eisai) can now be prescribed for IBD in Korea. Also, in addition to the JAK inhibitors, which were the only oral treatment options for severe IBD until the first half of this year, the launch and reimbursement approval of ‘Zeposia (ozanimod, BMS),’ a sphingosine-1-phosphate (S1P) receptor modulator broadened the options available on site. This means that the doctors have more options to use on patients who have failed initial treatment. In other words, the next treatment they choose can change the direction of each patient’s care. As such, the choice of IBD therapies, typified by ulcerative colitis (UC), has risen as a hot topic in clinical practice. While clinical research has made it possible to customize treatment for each patient, it has also increased competition between pharmaceutical companies. A professor of gastroenterology at A University Hospital in Busan said, "Previously, there were no options for IBD other than anti-TNF inhibitors. But many more options have become available in recent years, making the situation more complicated for prescribing clinicians.” The pharmaceutical industry is also scrambling to prioritize the lines of their treatment amid the various options becoming available. One representative example is BMS's Zeposia, which entered into a co-marketing agreement with Yuhan Corp, which owns strong domestic sales and marketing capabilities. An industry official said, "In fact, Yuhan Corp’s co-marketing agreement with BMS for Zeposia was considered quite unusual in the field. The collaboration seems to fall in line with the current situation, as the IBD market has recently become more competitive with an increasing number of treatment options and Yuhan had a need to increase its drug lineup." Also, with the increasing number of therapies available from global pharmaceutical companies, "sequencing" between existing and new drugs has become a hot topic in clinical practice. In other words, the increase in treatment options to use after initial treatment failure has created a "dilemma" as patients are allowed to switch between drugs, but not immediately. Another professor of gastroenterology at a university hospital said, "Recently, JAK inhibitors and small molecule drugs have been approved for ulcerative colitis and are being used in practice. However, the issue of side effects needs to be considered as well as benefits. Because complications such as herpes zoster and blood clots can occur when using these drugs, the new drugs are more commonly used on patients that have received shingles vaccinations and relatively young patients." He added, “In the JAK inhibitor class, Jyseleca is the only JAK inhibitor that can be prescribed directly after azathioprine. In clinical studies, RInvoq has shown promise in ulcerative colitis and Crohn's disease. However, the use of other JAK inhibitors requires azathioprine to be first removed, which is problematic for physicians because they have to remove azathioprine, which they consider effective for the patients.” In other words, while prioritizing the use of the right treatment for each patient, the sequencing of the treatments is currently being determined at the doctors’ discretion based on clinical research. As a result, therapies that are deemed to be the most effective based on the clinical studies that support their approval and reimbursement are being placed in the "last line of defense" and used as a last resort in practice. However, there is an opinion that this approach is not optimal and that prescriptions should be based on a comprehensive analysis of the patient's disease severity and socioeconomic status. There are also those who believe that they should wait and refer to the revisions made to the guidelines published by the Korean Association for the Study of Intestinal Diseases, which specializes in the disease. Professor Byong Duk Ye, Professor at the University of Ulsan College and Treasurer of KASID, said, "This is always a problem because there is no right answer. Especially, in the case of Rinvoq, there are opinions that its use should be delayed as much as possible because it is superior in clinical or endoscopic aspects compared to other agents. The drug should be used in combination with the patient's personal disease status and socioeconomic status." Ye added, "It is not necessarily a good treatment strategy to postpone the use of Rinvoq because it becomes less effective when used in later stages of the disease. It may be better to use it earlier to control inflammation. The final decision should be made in consultation with the patient, and we plan to publish revised KASID guidelines regarding the increased number of IBD treatment options available.”

- Policy
- Approval for PH-ILD inhalation drug 'Tyvaso' expected soon
- by Lee, Hye-Kyung Jun 11, 2024 05:48am
- Tyvaso Inhalation Solution. The approval of 'Tyvaso Inhalation Solution 0.6 mg/mL (treprostinil)' in South Korea is expected soon. It is approved in the United States as the treatment for pulmonary arterial hypertension (PAH) and pulmonary hypertension associated with interstitial lung disease (PH-ILD). According to industry sources on the 11th, the safety and effectiveness evaluation of Anterogen for the approval had been completed. The completion of the safety and efficacy evaluation is followed by the NDA. United Therapeutics’s Tyvaso is a treatment for patients with PH-ILD, and Anterogen has a domestic license. In August last year, the Ministry of Food and Drug Safety (MFDS) designated the drug as the 14th Global Innovative products on Fast Track (GIFT). Following the designation, Tyvaso has been under expedited review for approval. For GIFT-designated products, the review duration is shortened by 25% (i.e., 120→90 working days), rolling review is applied, which enables review on prepared and available documents, close communication between the reviewer and the developing company, such as product briefing and explanation of supplementation, is offered, and various regulatory supports, including RA consulting for fast commercialization, are provided. The company has applied for the efficacy and effectiveness of Tyvaso in PH-ILD, which is a fatal disease with no available treatments despite a survival rate of about 30% within three years of diagnosis. Tyvaso first received approval from the U.S. FDA in 2009 and received expanded indications for the first treatment of PH-ILD in 2021. The approval was based on the INCREASE clinical trial data. The trial was the largest, enrolling 326 patients with PH-ILD and the most comprehensive. Tyvaso met the primary endpoint and significantly improved 6-minute walk distance (6MWD) results. The treatment showed benefits in various major subgroups related to etiology, disease severity, age, sex, blood kinetics, and doses. Improvements in the secondary endpoints included the reduction of NT-proBNP levels, a heart biomarker, the time to clinical worsening, the change in the highest 6MWD at week 12, and the change in the lowest 6MWD at week 15.
- Company
- GC's Sanfilippo syndrome drug receives fast-track status
- by Son, Hyung-Min Jun 11, 2024 05:47am
- GC Biopharma announced on Tuesday that the U.S. FDA has granted Fast Track Designation for GC1130A, a treatment for Sanfilippo syndrome type A (MPS IIIA) that it has been co-developing with Novel Pharma. The fast-track designation follows the FDA's clearance of the Phase I investigational new drug (IND) application for GC1130A last month and is expected to further accelerate the development of GC1130A. Sanfilippo syndrome (type A) is a rare genetic disorder that causes central nervous system damage through the accumulation of heparan sulfate, leading to progressive neurological decline. Without treatment, patients face life-threatening complications by the age of 15. GC1130A is a new biological drug that is being developed using GC Biopharma’s high-concentration protein formulation technology, designed for administration to the central nervous system. It is delivered directly into the brain's ventricles through intracerebroventricular (ICV) injection, a method first applied globally by GC Biopharma's Hunter syndrome treatment, 'Hunterase', which has received marketing authorization in Japan. The potential of GC1130A to meet the unmet medical needs of Sanfilippo syndrome has been recognized by major drug regulatory agencies; in 2023, the FDA granted GC1130A Rare Pediatric Disease Designation (RPDD) and Orphan Drug Disease (ODD), and earlier this year, the European Medicines Agency (EMA) also granted GC1130A ODD status. Currently, GC Biopharma and Novel Pharma are preparing to initiate a multinational clinical trial to evaluate the safety and tolerability of GC1130A in Korea, the US, and Japan. A GC Biopharma official said, “With no approved treatment available for Sanfilippo syndrome, we are pleased that the FDA has granted GC1130A Fast Track designation. This designation will allow us to accelerate the development of our drug to bring hope to patients suffering from Sanfilippo syndrome.” The FDA's Fast Track program is designed to expedite the development and review of drugs intended to treat serious or unmet medical needs. The Fast Track designation provides extensive support, including frequent meetings with the FDA throughout the drug development, clinical, and approval stages.
- Policy
- Handok paces its efforts to reimburse Pivlaz in KOR
- by Lee, Tak-Sun Jun 11, 2024 05:47am
- Handok is taking a break from its pursuit of reimbursement for ‘Pivlaz,’ a new drug used to prevent cerebral vasospasm in patients with subarachnoid hemorrhage that the company is supplying and distributing in Korea. The company immediately applied for Pivlaz’s reimbursement to the Health Insurance Review and Assessment Service after receiving approval in December last year, but was found to have voluntarily withdrawn the application recently. The company is expected to realign its strategy and then pursue reimbursement again. According to industry sources on the 10th, Handok submitted a letter requesting the withdrawal of the application for the drug determination (reimbursement) of Pivlaz Inj (clazosentan). The drug was granted marketing authorization last year on December 7. It is a selective endothelin A receptor antagonist indicated for the prevention of cerebral vasospasm, vasospasm-related cerebral infarction, and cerebral ischemic events in adults who have undergone clipping surgery or coiling treatment for aneurysmal subarachnoid hemorrhage. Pivlaz is the first drug approved in Korea to prevent both cerebral vasospasm and its complications. Cerebral vasospasm, which occurs after aneurysmal subarachnoid hemorrhage, has been shown to double the risk of death in patients. It is also accompanied by serious complications such as localized paralysis, speech impairment, and decreased consciousness. Active prevention and treatment are crucial, but the field has been experiencing difficulties due to the lack of appropriate drugs. This is why Pivlaz is rising as a promising treatment option. Nxera Pharma Korea owns the sales rights for Pivlaz in Korea. The Japanese multinational company, former Sosei Heptares, changed its name to Nxera in April last year after acquiring the Swiss pharmaceutical company Idorsia, which developed Pivlaz. On April 12, Handok announced that it signed an exclusive domestic supply and distribution agreement with Nxera Pharma Korea for Pivlaz. Handok has been collaborating with Nxera Pharma Korea Korea since 2008 to conduct domestic clinical trials and obtain marketing authorization for Pivlaz. Handok and Nxera Pharma Korea are seeking to launch Pivlaz early next year. There is still time to receive reimbursement within the timeline. The industry’s attention is focused on whether the two companies will be able to realign their strategy and speed up the reimbursement process for Pivlaz.
- Company
- 'Novel drugs·CDMO competitiveness↑'
- by Son, Hyung-Min Jun 10, 2024 05:41am
- Major biotech companies in Korea participated in the BIO International Convention (BIO USA 2024) and introduced their in-house competitiveness. Over 50 Korea-based companies attended the BIO USA 2024, held between June 3-6 in San Diego, United States. They sought opportunities to expand partnerships along with discussions for technology transport. Samsung Biologics showcased its new platform, showing a strong directive to expand the company’s growth related to intensified cell-culture with improved manufacturing capacity. Lotte Biologics, ST Pharm, and Prestige Biologic introduced their contract development and manufacturing organization (CDMO) capacities and held partnering meetings for contract orders. Korean companies have made significant achievements in novel drug development. VaxCell Biotherapeutics showcased positive results of its novel candidate under a phase 2 clinical trial for hepatocellular carcinoma. Genome & Company won a technology transport contract for its antibody-drug conjugate (ADC), confirming its competitiveness. China absent from the BIO USA…all eyes on K-bio According to industry sources on June 9th, 47 Korean companies ran booths during the BIO USA, which was 6 more companies than last year. BIO USA is the world’s largest biopharmaceutical convention, with more than 20,000 leaders in the biopharmaceutical industry participating. Continuing from last year, Korean CDMO companies have gathered attention this year. Notably, due to the introduction of the Biosecure Act by the United States, all eyes were drawn to the Korean companies in the absence of WuXi Biologics, China’s largest CDMO. The United States aims to limit the business with the Chinese biotech companies. Therefore, the analysis suggests that along with Swiss-based Lonza and Japan-based Fujifilm, Korean CDMO companies will have more opportunities. The Korea booth at the BIO International Convention (BIO USA 2024) (photo=KoreaBio). Samsung Biologics launched a new CDO platform called S-Tensify, bolstering CDO competitiveness. S-Tensify supports high-intensity biomedicine development with the latest cell-culture technology. The company shared that S-Tensify’s adoption of N-1 perfusion technology has increased its cell-culture concentration by 30-fold, significantly boosting manufacturing capacity during inoculation at the seed stage (N-1). ST Pharm has started CDMO of CRISPR/Casx, used in gene editing technology. Gene editing is a biomolecular tool for editing precise locations within DNA using zinc finger nucleases, TALEN, or CRISPR/Cas9. CRISPR/Casx is an innovative technology for precisely editing DNA sequences, enabling deletion, addition, or correction of genetic material. Novel drug development using gene editing is currently underway. The U.S. FDA approved exa-cel (U.K.-authorized product name: Casgevy), a CRISPR/Cas9 gene-edited therapy developed by the United States biotech company Vertex Pharmaceuticals and SWISS-based CRISPR Therapeutics. ST Pharm showcased its manufacturing process of sgRNA, which precisely targets the genome. More than 100 mer high-purity sgRNA production is needed to develop medicines, and this manufacturing technology is more difficult than ASO or siRNA oligonucleotides ingredients. During the event, Prestige Biologics shared new CDMO business opportunities due to the imminent passing of the Biosecure Act by the United States. According to the company, over 30 companies have requested meetings, and 6 offers have been received. Prestige Biologics emphasized that the company is equipped with cost competitiveness and quality for a single-use approach. K-bio showcased novel drug development competitiveness In addition to CDMO orders, Korean biopharmaceutical companies also showcased their capabilities in the development of novel drugs. VaxCell Biotherapeutics presented the outcomes of Phase 2a ‘Vax-NK/HCC’ clinical trials for hepatocellular carcinoma. This trial evaluated the efficacy of Vax-NK/HCC plus HAIC combination therapy involving 16 patients with advanced hepatocellular carcinoma who did not respond to conventional treatments. The clinical results demonstrated that Vax-NK/HCC plus HAIC combination therapy had an objective response rate (ORR) of 68.8% and a progression-free survival (PFS) of 16.8 months. Genome & Company has confirmed the competitiveness of its ADC candidate. Earlier this month, the company signed a technology transfer contract with Swiss-based Debiopharm for its ADC candidate, GENA-111. Including the upfront payment of approximately KRW 6.9 billion, the contract size amounts to a maximum of KRW 586.4 billion. GENA-11 is an ADC candidate with a novel mechanism for targeting CD239. CD239 is known to be highly expressed in cancer cells than in healthy cells, and no ADC has been commercialized for targeting this. Genome & Company is considering targeting GENA-111 to treat gynecologic cancer. After assessing which payload would be more effective, the company stated that the final target indication would be decided. Bridge Biotherapeutics shared directives for developing novel drug candidates to treat idiopathic pulmonary fibrosis (IPF). BBT-887 is an innovative new candidate under development in a phase 2 trial. It is a selective inhibitor of autotaxine enzyme. Autotaxine is a protein known to bind to receptors in cells and induce various physiological activities, such as sclerosis and tumorigenesis. Bridge Biotherapeutics presentation (photo=Bridge Biotherapeutics). Last month, Bridge Biotherapeutics received authorization from the Independent Data Monitoring Committee (IDMC) to continue the clinical trial. After evaluating the efficacy and safety data of 75 clinical subjects, there were no concerns related to the drug safety or effects.
- Product
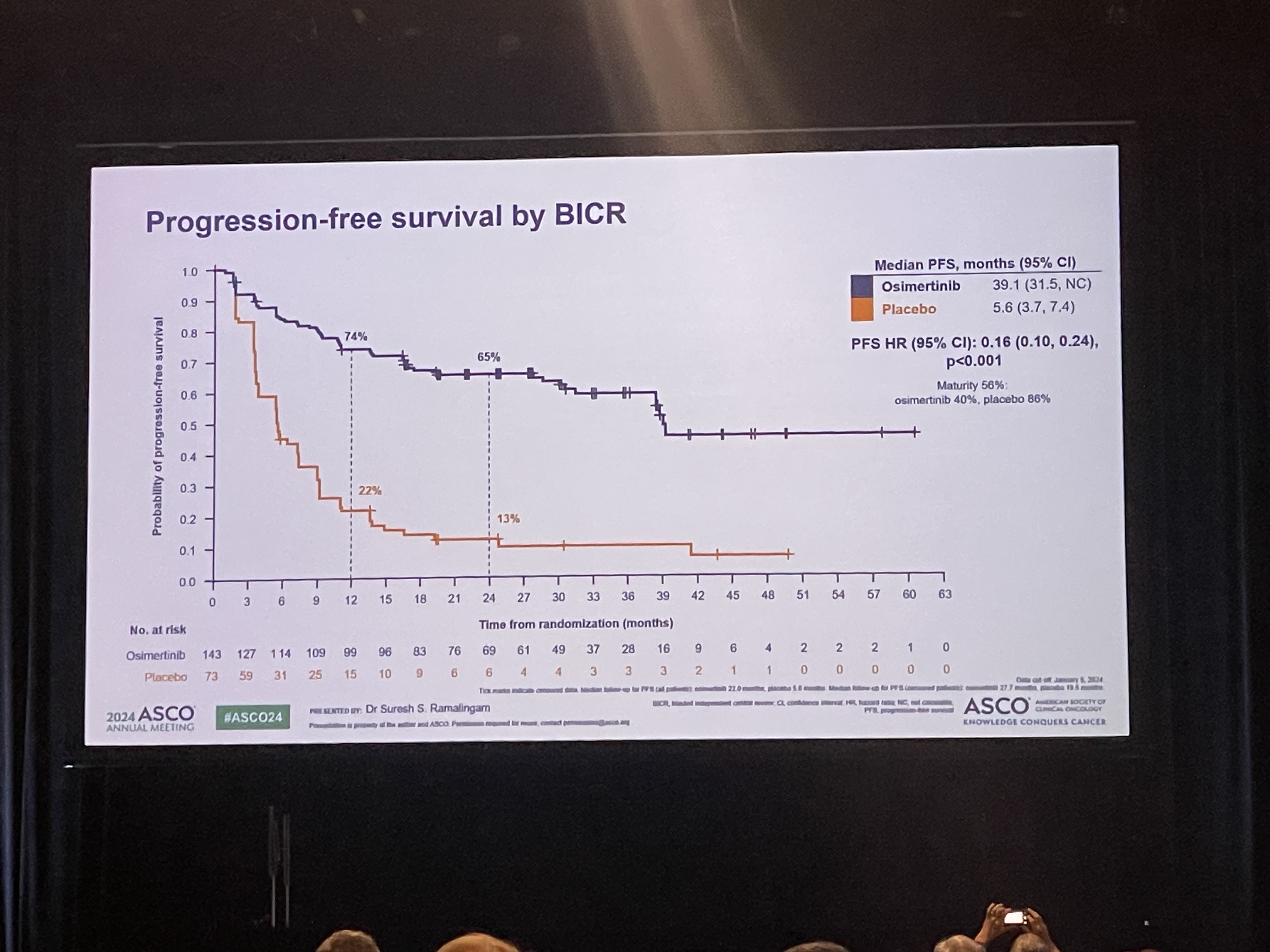
- Will Osimertinib emerge as the standard of care
- by Park, sang-jun Jun 10, 2024 05:41am
- The LAURA trial, which evaluated osimertinib’s effect in patients with unresectable Stage III EGFR-mutant non-small cell lung cancer who received chemoradiotherapy (CRT), was presented at the 2024 American Society of Clinical Oncology (ASCO) Annual Meeting. The results were also concurrently published in NEJM. # The LAURA trial evaluated progression-free survival (PFS) in 216 patients with unresectable stage III EGFR-mutant NSCLC who received chemoradiotherapy (CRT). The patients were randomized to receive osimertinib or placebo. The results showed a median PFS of 39.1 months and 5.6 months in the osimertinib and placebo arms, respectively, with an 84% reduction in the risk of disease progression and death in the osimertinib arm. The overwhelming numbers were met with spontaneous resounding ovation. Although the overall survival rates were not clear yet, researchers also added a positive interpretation based on the fact that overall survival did show a clear trend toward improved survival in the osimertinib arm, even though 80% of the placebo arm switched to osimertinib. Professor Suresh S. Ramalingam from the Winship Cancer Institute at Emory University School of Medicine, who presented results of the phase III LAURA study during the Plenary Session at the 2024 ASCO Annual Meeting, said, “The current standard of care for unresectable stage III EGFR-mutant NSCLC patients following CRT is durvalumab, but the benefit of the immunotherapy agent, specifically among patients with EGFR mutations, is uncertain. Based on the clear benefits, osimertinib after CRT will most likely emerge as the new standard of care for EGFR-mutant disease in this setting.” The next big question will be in setting the eligible subjects and timing of administration. Patients with unresectable stage III EGFR-mutant NSCLC who have received chemoradiotherapy (CRT) are regarded as an incurable group of patients, who have a high likelihood of relapse in the future. This is why drug use in this patient group needs to be reviewed from various aspects. Professor Lecia V. Sequist from the Massachusetts General Hospital and Harvard Medical School, who attended the presentation as a discussant for the abstract, regarded the results as a half glass of water, explaining that osimertinib may and may not be a viable treatment option depending on the perspective.” He emphasized that the positive benefits of osimertinib in terms of preventing brain metastases are a clear advantage, but the cost of the drug and increased side effects are a disadvantage. Professor Beung-Chul Ahn of the National Cancer Center, said, "The positive outcome of osimertinib in this group of patients is very welcome evidence, but if we evaluate it soberly, the cost of the drug cannot be ignored in clinical practice, and there are groups of patients who do not necessarily need it, so we need a treatment strategy that reviews its use according to the situation.”