- LOGIN
- MemberShip
- 2026-05-05 15:11:19
- Company
- Discussions on expanding benefit for Jakavi's GVHD are slow
- by Eo, Yun-Ho Jun 13, 2023 05:43am
- Discussions on expanding insurance coverage for Jakavi's Graft-versus-Host Disease (GvHD) indication have been sluggish. According to the industry, Novartis Korea Jakavi passed the harmaceutical Reimbursement Criteria Subcommittee of the Health Insurance Review and Assessment Service last February, but it has not been submitted to the Financial Impact Assessment Subcommittee, which is a step before the Pharmaceutical Reimbursement Evaluation Committee. In May 2022, after obtaining indications for graft-versus-host disease in Korea, the drug submitted an application for an extension of coverage. However, after being put on hold for more than 8 months, discussions on salary have begun. GvHD is a serious complication that can occur after an allogeneic hematopoietic stem cell transplant (allo-SCT). The transplanted donor's T cells recognize the patient's normal cells as foreign substances and attack them, affecting various organs such as the skin, gastrointestinal tract, liver, and lungs. As symptoms can appear throughout the body, GvHD poses another challenge to patients who have survived allogeneic hematopoietic stem cell transplantation and affects their quality of life. Steroids are used as the first-line therapy, but about 50% of them fail, and in these cases, standard treatment has not yet been established, so there is an unmet demand for effective treatment options. Jakavi is an option for the treatment of patients with acute or chronic graft-versus-host disease aged 12 years or older who do not respond adequately to prior corticosteroid therapy in these circumstances. Kim Hee-jae, chairman of the Korean Society for Hematopoietic Stem Cell Transplantation (Professor of Hematology at Seoul St. Mary’s Hospital), said, “Jakavi has shown good effects through clinical studies in the treatment of acute and chronic patients, and similar results have been shown in actual practice. “It opens up new possibilities for many patients,” he said. Jakavi has proven its efficacy in GvHD through the phase 3 REACH2 study. As a result, the overall response (OR) on day 28 of the Jakavi-administered group was 62% (96/154), which was higher than that of the control group, Best Available Therapy (BAT)-administered group, 39% (61/155). On the 56th day, the durable overall response was confirmed to be 40% (61/154), about twice as high as 22% (34/155) of the control group. More about this source texture text is required for additional translation information.
- Company
- Severe asthma Tx Tezpire to soon be commercialized in Korea
- by Eo, Yun-Ho Jun 13, 2023 05:43am
- The new drug for severe asthma, ‘Tezpire' is soon to enter the Korean market. According to industry sources, AstraZeneca Korea has submitted an application for the approval of ‘Tezspire (tezepelumab)’ in Q2 to the Ministry of Food and Drug Safety and is receiving review. At this pace, Tezspire is expected to be commercialized in the second half of the year. As a viable competitor to Sanofi’s ‘Dupixent (dupilumab),’ Tezspire inhibits the action of the thymic stromal lymphopoietin (TSLP), a key epithelial cytokine that sits at the top of multiple inflammatory cascades, to block the inflammatory chain reaction. The drug was approved by the US FDA in 2021 as a treatment for adult and pediatric patients aged 12 years and older with severe asthma., and added a self-injection formulation to its approval in February this year. Tezspire demonstrated a significant reduction in asthma exacerbation in the Phase II PATHWAY trial and Phase III NAVIGATOR trial, which included a broad population of severe asthma patients irrespective of key biomarkers, including blood eosinophil counts, allergic status, and fractional exhaled nitric oxide (FeNO). The most common adverse reactions shown in those that received Tezspire in the trials were pharyngitis, rash, arthralgia, and injection site reactions. The findings from the NAVIGATOR study were previously published in The New England Journal of Medicine. The Korea National Enterprise for Clinical Trials had selected Tezspire as the No.1 drug in need of an urgent introduction to Korea in its report on the ‘List of foreign new unintroduced drugs that should be promptly Introduced to Korea.’
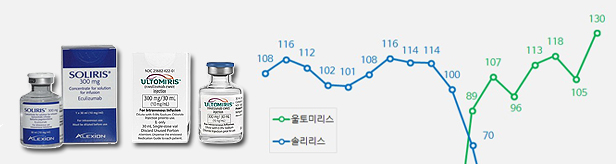
- Company
- Sales of Soliris and Ultomiris rise despite changes
- by Kim, Jin-Gu Jun 13, 2023 05:43am
- Soliris(left) and Ultromiris Quarterly sales of the rare disease treatments ‘Soliris (eculizumab)’ and ‘Ultomiris (ravulizumab)’ had risen significantly in Q1 this year. Although sales of Soliris remained similar to last year, sales of Ultomiris, its follow-on drug, soared in the same period. The analysis is that the generation change between the two drugs has neared completion. According to the market research institution IQVIA on the 12th, sales of Ultomiris in Q1 were KRW 13 billion, up 35% from KRW 9.6 billion in Q1 last year. During the same period, Soliris’s sales slightly fell from KRW 2.8 billion to KRW 2.4 billion. Soliris is a rare disease treatment indicated for the treatment of ▲ paroxysmal nocturnal hemoglobinuria, ▲ atypical hemolytic uremic syndrome (aHUS), ▲generalized myasthenia gravis, and ▲ neuromyelitis optica spectrum disease. Ultomiris is a follow-on of Soliris. As it has a half-life that is 4 times longer than that of Soliris, the drug may be administered every 8 weeks. However, the indication for the drug is limited to paroxysmal nocturnal hemoglobinuria/ Both drugs were introduced to Korea by Handok from Alexion. Soliris was approved in January 2010 and applied for reimbursement in 2018. Ultomiris was approved in May 2020 and its reimbursement was applied from June 2021. The combined sales of the two drugs peaked at KRW 15.9 billion in Q3 2021 and then was on a gradual decline. In Q4 last year, its sales decreased to KRW 12.9 billion. However, sales showed a rebound in Q1 this year, recording combined sales of KRW 15.4 billion. Quarterly Sales of Soliris·Ultromiris (Unit: KRW 100 mil, Data: IQVIA) The sales rebound received more attention because the domestic sales rights for the two drugs were transferred from Handok to AstraZeneca in Q1 last year. AstraZeneca acquired Alexion in 2020 for USD 39 billion (approximately KRW 42 trillion). After the sales rights agreement between Handok and Alexion expired in January this year, AstraZeneca Korea retrieved the sales rights to sell the two rare disease drugs directly in Korea. However, the prevailing opinion in the industry is that the sales right transfer had little or limited effect on the sales rebound. AstraZeneca established a rare disease Business Unit at the end of last year and completed the composition of the business unit in February of this year in time for the retrieval of Soliris and Ultomiris’s sales rights. The company was able to start full-scale marketing activities only in March. Therefore, the sales rebound could be rather interpreted as performance derived under the strong influence of Handok. Industry experts believe that the impact of the sales right transfer would only be measurable after Q2.
- Company
- Beova can be prescribed at general hospitals
- by Eo, Yun-Ho Jun 12, 2023 05:42am
- Beova, a new drug introduced by Jeil Pharmaceutical, will be available for Rx at general hospitals. According to related industries, Beova, an overactive bladder treatment, is currently available at 22 medical institutions nationwide, including SMC and AMC, as well as advanced general hospitals, Gangnam Severance Hospital, Sangnam Sacred Heart Hospital, Dongtan Sacred Heart Hospital, Chungnam National University Hospital, and Hanyang University Hospital (headquarters, Guri). Beova is a new drug introduced by Jeil Pharm from Japan's Kyorin Pharmaceuticals in November 2019. It obtained domestic development, manufacturing, and sales rights, and imported raw materials to produce finished products in domestic factories. In order to obtain domestic approval, Jeil Pharmaceutical conducted a phase 3 bridging clinical trial to compare and evaluate the safety and effectiveness of Beova in 210 overactive bladder patients at 20 domestic institutions, including Seoul Asan Medical Center, from May 2020 to this year. As a result, it was confirmed that the change in average urination frequency per day at 12 weeks from baseline as the primary efficacy evaluation variable was improved by -2.38 times compared to -1.22 times with a placebo. In the secondary endpoints, the average number of urinary urgency per day, the number of urge incontinence, the change in the number of urinary incontinence, and the change in the average voiding volume per time, there was a significant improvement compared to the placebo. Beova is a new β3-adrenergic receptor agonist that selectively acts on sympathetic nerve receptors to relax the bladder detrusor muscle, helping to treat symptoms such as frequent urination, urgency, and urgency urinary incontinence. Mirabegron was the only β3-adrenergic receptor agonist currently on the market, but the launch of Beova is expected to form a competitive landscape. Vibegron is known to be more than 9000 times more selective for β3 receptors than β1 and β2 receptors. In addition, Vibegron's maximum response rate for β3 receptors is 99.2%, which is higher than 80.4% for Mirabegron, the same β3 agonist. It is an ingredient that has the advantage of having a low risk of cardiovascular side effects because it does not stimulate receptors. It is known that there is very little concern about interactions with drugs that pass through the CYP2D6 metabolic pathway and that it can be administered in normal doses even to patients with hepatic or renal impairment. Lee Gyu-seong, professor of urology at SMC, said, "Beova is a new drug that is introduced in Korea. It will help provide high-quality treatment effects to overactive bladder patients in Korea with excellent symptom improvement and low adverse reaction rates."
- Company
- Eisai applies for approval of its AD drug lecanemab in KOR
- by Jung, Sae-Im Jun 12, 2023 05:42am
- Pic of lecanemab The drug that is being considered the next blockbuster candidate, Eisai and Biogen’s Alzheimer’s treatment ‘lecanemab,’ is set to enter the Korean market. Eisai announced on the 8th that it had submitted an application for the marketing authorization of lecanemab to the Ministry of Food and Drug Safety to treat mild cognitive impairment or mild dementia stage of disease arising from Alzheimer’s disease (AD). This is the third application the company had filed in Asia after Japan and China. Lecanemab was jointly developed by the two companies. Lecanemab selectively binds to neutralize and eliminate soluble, toxic amyloid-beta (Aβ) aggregates (protofibrils) that are thought to contribute to the neurodegenerative process in AD. The drug received accelerated approval from the U.S. Food and Drug Administration in January. As Eisai applied for formal approval, the FDA plans to hold an advisory committee on the 9th (US local time) and decide whether to grant approval next month. Unlike 'Aduhelm (ingredient: aducanumab)', which failed commercialization, high expectations are in place for lecanemab’s growth into a blockbuster drug. Clarivate, a global academic information service company, predicted that lecanemab’s sales would reach USD 1.02 billion (KRW 1.32 trillion) in 2027 in its ‘Drugs to Watch’ report that it released earlier this year. Eisai’s application to the MFDS is based on the Clarity AD Phase III and Phase IIb clinical studies that confirmed that lecanemab reduced clinical cognitive decline in early Alzheimer's disease. The Phase III Clarity AD compared lecanemab with placebo in 1.705 patients aged between 50 to 90 with early AD. The primary efficacy endpoint was the change in clinical decline on the global cognitive and functional scale, CDR-SB, compared with placebo at 18 months. Key secondary endpoints were ▲the AD Assessment Scale-cognitive subscale14 (ADAS-cog14), ▲change from baseline at 18 months compared with placebo of treatment in amyloid levels in the brain measured by amyloid positron emission tomography (PET), ▲AD Composite Score (ADCOMS), etc. Change in CDR-SB score with lecanemab -placebo (primary endpoint) (data: Biogen) Results showed that the lecanemab arm recorded a CDR-SB score of 1.21 at 18 months, which is a 27% reduction in clinical decline compared with the 1.66 recorded in the placebo arm. This delay started as early as six months. The drug also achieved statistically significant in its key secondary endpoints compared with placebo. In the amyloid PET subgroup analysis, the lecanemab arm showed a statistically significant reduction in brain amyloid accumulation from 3 months. The ADAS-Cog14 evaluation results showed that cognitive decline was delayed by 26% with lecanemab. ADCOMS results also showed a 24% delay in disease progression at 18 months with lecanemab.
- Company
- Fidanacogene Elaparvovec designated as an orphan drug
- by Eo, Yun-Ho Jun 12, 2023 05:42am
- Fidanacogene Elaparvovec, a one-shot hemophilia treatment, has been designated an orphan drug. The Ministry of Food and Drug Safety recently announced that it has selected Fidanacogene Elaparvovec, Pfizer's hemophilia B gene therapy, as an orphan drug. Fidanacogene Elaparvovec is a method that combines adenoviral vector (AVV) capsid and highly active coagulation factor IX gene and is characterized by producing coagulation factor IX in one cycle instead of regular injection am. The drug has been designated as a Breakthrough Therapy, Advanced Regenerative Medicine and Therapeutics (RMAT), and Orphan Drug by the US FDA, and PRIority MEdicines and Orphan Drug by the European EMA. The phase 3 BENEGENE-2 study confirming the effectiveness of Fidanacogene Elaparvovec also received significant attention. The study evaluated the efficacy and safety of Fidanacogene Elaparvovec in patients with a factor 9 of 2% or less, and the participating patients were evaluated for 6 years based on a single intravenous injection. The main purpose of this study is to determine how much gene therapy reduces ABR compared to SOC. According to the topline results released recently, the Fidanacogene Elaparvovec group met the primary endpoint by demonstrating non-inferiority and superiority compared to standard therapy in ABR. From 12 weeks to 15 months, the average ABR of the Fidanacogene Elaparvovec group was 1.3, whereas that of the standard therapy group was 4.43. Gene therapy reduced ABR by 71%, confirming its superiority over standard therapy. The main secondary endpoint was the ABR measured based on treatment. The Fidanacogene Elaparvovec group reduced treatment-based ABRs by 78% and annual infusions by 92% compared to the standard-of-care group. The Ministry of Food and Drug Safety is operating an orphan drug designation system to support the development of treatments for rare and incurable diseases. Among drugs used for the purpose of diagnosing or treating rare diseases, drugs that cannot be replaced or that are significantly improved over drugs that can be replaced may be designated as orphan drugs. If designated as an orphan drug, you can receive benefits such as being subject to expedited review at the time of product approval.
- Company
- Chong Kun Dang hastens way into diabetes Tx market
- by Kim, Jin-Gu Jun 12, 2023 05:42am
- Chong Kun Dang is working to accelerate the expansion of its diabetes treatment portfolio. In addition to its own ‘Duvie (lobeglitazone),’ the company received approval for diabetes combination drugs that contain 'Januvia (sitagliptin),’ which it had acquired rights for in Korea. The release of such combination drugs is considered the company’s strategy to preoccupy the market before the patent expiry of Januvia. According to industry sources on the 12th, Chong Kung Dang received approval for its Duvie Tab from the Ministry of Food and Drug Safety on the 9th. The drug is a combination between sitagliptin and lobeglitazone. The company had received approval for Duvie, a TZD-class diabetes treatment in 2013 as the 20th new new homegrown drug in Korea. In May, it acquired the license for MSD's DPP-4 inhibitor class diabetes treatment, Januvia. It acquired all rights, including domestic sales, distribution, licensing, trademark, and manufacturing, for not only Januvia but also Janumet and Janumet XR. As a result, Chong Kun Dang now owns 2 original drugs in the diabetes market. Immediately after acquiring Januvia, Chong Kun Dang obtained approval for a series of combination drugs that contain lobeglitazone and sitagliptin. On the 2nd of last month, Chong Kun Dang obtained approval for DuviMet-S XR, a three-drug combination for diabetes consisting of lobeglitazone, sitagliptin, and metformin. With the addition of Duvie-S to the lot, Chong Kun Dang's Duvie-based diabetes lineup has increased to amount to a total of 4: Duvie Tab which was approved in 2013, DuviMet XR Tab that was approved in 2016 (lobeglitazone + metformin), and DuviMet-S SR Tab and Duvie S Tab that were added this year. The company is expected to continue expanding its diabetes treatment portfolio for some time. Chong Kun Dang is currently conducting 4 clinical trials to treat diabetes: Phase III clinical trials for CKD-383, CKD-398, and CKD-371 and Phase I clinical trials for CKD-379. Among them, CKD-383 and CKD-379 are three-drug combination drugs for diabetes, presumably a combination product based on Duvie or Januvia. Chong Kun Dang The industry predicted that the imminent expiry of Januvia's patent was behind Chong Kun Dang's rapid expansion of its diabetes portfolio. Januvia's patent is set to expire in September. About 100 companies, excluding Chong Kun Dang and MSD, are expected to simultaneously release single and combination drugs that contain sitagliptin at the time of patent expiry. With such fierce competition being expected, Chong Kun Dang’s move is interpreted as a strategy to preoccupy the market by releasing a combination drug based on sitagliptin and lobeglitazone 2-3 months in advance. In particular, in a situation where the growth of both its Duvie series and the Januvia series are have been slowing down, attention is focused on how much synergy the combination of the two drugs will bring to the market. According to the market research institution UBIST, Duvie and Duvimet recorded a combined prescription amount of KRW 25.4 billion last year. This was a slight increase from the KRW 25.1 billion it made in 2021. In Q1 this year, the drugs recorded KRW 6.1 billion, down KRW 100 million from the previous year. The Januvia series recorded prescription sales of KRW 162.5 billion last year. The amount decreased by 8% compared to the KRW 176.3 billion the series had made in 2021. In Q1 this year, prescription sales were KRW 37.9 billion, down 9% from the previous year.
- Company
- Crystal Genomics supplies 2.6 billion Acelex to Russia
- by Lee, Seok-Jun Jun 09, 2023 05:36am
- Crystal Genomics will supply $2 million of Acelex, an osteoarthritis drug, to Russia. According to the company on the 7th, this order is based on a supply contract with PharmArtis International, a Russian state-run pharmaceutical company. This is the second order quantity. The contractual obligation to purchase is $43.86 million. Crystal Genomics will receive up to $77.6 million in milestones based on additional sales. PharmArtis is currently developing sales and marketing strategies and more. It will officially release Acelex in Russia next month at the earliest. The Russian COX-2 inhibitor market, to which Acelex belongs, is showing an annual growth rate of more than 30%. It is possible to expand the market to neighboring Eurasian Economic Community regions (Belarus, Kazakhstan, Armenia, Kyrgyzstan, etc.). The Eurasian Economic Community regional regulatory authorities are in the process of obtaining Acelex approval. Acelex is Korea's first bio-venture new drug. It selectively inhibits only COX-2 among cyclooxygenases (COX-1 and COX-2), two enzymes that act on the formation of prostaglandins that cause inflammation and pain. It was developed to overcome stomach cramps and gastrointestinal disorders, which are side effects of non-steroidal anti-inflammatory drugs. An official from Crystal Genomics said, "Following the shipment of the first volume at the end of March, we received an order for the second volume, so local sales are imminent. Sales in Russia are expected to improve performance."
- Company
- Korea’s new drug reimb rate is below OECD average
- by Eo, Yun-Ho Jun 09, 2023 05:35am
- A survey found that Korea’s new drug reimbursement rate does not amount to the OECD average. The Korean Research-based Pharmaceutical Industry Association (KRPIA, Chairman: Dong-Wook Oh) recently announced Korea’s new drug release status based on its ‘Global Access to New Medicines Report’ According to the report, it takes longer than the Organisation for Economic Co-operation and Development (OECD) country average for new drugs to be introduced to Korea after its global launch, and Korea’s release rate and reimbursement rate were also below the OECD country average. The report was based on the ‘Pharmaceutical Research and Manufacturers of America’ report that was published in April. PhRMA’s report subdivided 72 countries including Korea by G20, OECD, and region, and investigated the current status of new drug launches and health insurance coverage in each country. The report was prepared based on an investigation of a total of 460 new drugs that have received marketing authorization in the United States, Europe, and Japan over the past 10 years from 2012 to 2021. When taking a closer look, the report showed that the ratio of new drugs introduced to Korea within 1 year after the initial global launch was found to be less than half of the OECD average. While the average new drug introduction rate (non-reimbursement release rate) in OECD countries is 18%, Korea's level was only 5%, about a quarter of the OECD average. The countries with the highest new drug introduction rates were the United States (78%), Germany (44%), then the United Kingdom (38%). Also, Japan’s new drug introduction rate was nearly twice that of Korea, at 32%. By disease, it took about 27 to 30 months for anticancer drugs and new rare disease drugs to be released in Korea without reimbursement after their global launch. Compared to the average of 12 to 15 months it took in other advanced countries such as the UK and Germany and 18 to 21 months in Japan, it takes twice as long for new drugs to be launched in Korea. Moreover, it took a total of 46 months from the initial global launch of a new drug to reimbursement in Korea. The average time taken in other OECD countries was similar at 45 months, but compared to Japan (17 months) and France (34 months), it took from 10 months to more than twice longer for drugs to be reimbursed in Korea. Meanwhile, the share of new drugs covered by health insurance in Korea was 22%, which was below the OECD average (29%). This is only half that of Japan (48%) and the UK (48%). KRPIA said, “The report holds significance for allowing us to identify the rate and period of new drugs introduced and reimbursed in Korea after their global launch. Improvements should be made to Korea’s system and environment in advance to enable prompt reimbursement of global new drugs for patients in Korea.
- Company
- Non-covalent BTKi pirtobrutinib receives orphan drug desig
- by Eo, Yun-Ho Jun 09, 2023 05:35am
- The BTK inhibitor ‘pirtobrutinib‘ received an orphan drug designation in Korea. The Ministry of Food and Drug Safety recently announced that it had designated Lilly’s Bruton's Tyrosine Kinase (BTK) inhibitor Jaypirca (pirtobrutinib) as an orphan drug. The subject indication is as monotherapy for adult patients with relapsed or refractory mantle cell lymphoma who have been previously treated with a BTK inhibitor. The US FDA granted accelerated approval for Jaypirca in January. The drug received attention as the first non-covalent BTK inhibitor and as an alternative to patients who failed treatment with covalent BTK inhibitors like ‘Imbruvica (ibrutinib),’ ‘Brukinsa (zanubrutinib),’ etc. Jaypirca’s efficacy was evaluated through the BRUIN trial. Results showed that the overall response rate of patients that received 200mg of Jaypirca once a day was 50% (60 patients) among the 120 mantle cell lymphoma patients, with a complete response rate of 13% (15 patients) and a partial response rate of 38% (45 patients). The estimated median duration of response was 8.3 months, and the estimated duration of response rate at 6 months was 65.3%. The pooled safety analysis of the full BRUIN study population evaluated 583 patients with hematologic malignancies administered Jaypirca 200 mg daily as a single agent. In this pooled safety population, the most common adverse reactions (ARs) to Jaypirca therapy, occurring in 20% of patients or more, were decreased neutrophil count, decreased hemoglobin, decreased platelet count, fatigue, musculoskeletal pain, decreased lymphocyte count, bruising, and diarrhea. Meanwhile, the MFDS has been operating the orphan drug designation system to support the development of treatments for rare and intractable diseases. Among the drugs used to diagnose or treat rare diseases, drugs that are irreplaceable or those that significantly improved outcomes compared to their alternatives are designated as orphan drugs. Drugs designated as orphan drugs can receive benefits such as being eligible for accelerated review for marketing authorizations, etc.