- LOGIN
- MemberShip
- 2026-07-22 06:53:40
- Company
- Cochrane review on ineffectiveness of Alzheimer’s drug sparks backlash
- by Eo, Yun-Ho May 07, 2026 10:29am
- “Anti-amyloid Alzheimer’s drugs show no clinically meaningful effect.”The backlash is as fierce as the message itself. Last April, the Cochrane Database published the results of a systematic review and meta-analysis covering anti-amyloid targeted therapies.The study concluded that these drugs, as a whole, failed to demonstrate clinically meaningful benefits in inhibiting cognitive and functional decline and raised significant safety concerns, such as amyloid-related imaging abnormalities (ARIA).Cochrane is a nonprofit health research organization with over 11,000 experts from more than 190 countries worldwide, providing evidence to inform healthcare decision-making. Such an announcement in a prestigious academic journal naturally had a significant impact.However, before assessing the value and validity of the study, the academic community has come forward to directly refute the results of the Cochrane meta-analysis.It is true that the conclusions of a meta-analysis can vary completely depending on “how the data is pooled.”Medical professionals argue that the core of the controversy surrounding this review lies in the fatal “methodological limitations” inherent in the study design.Because the study produced results that fundamentally overturn the current landscape of clinical practice, it is also true that any misinterpretation could cause significant turmoil for patients with early-stage Alzheimer’s disease, whether they are about to begin treatment or are already undergoing it, as well as their families.Integration of drugs with different development stages and regulatory statusThe main criticism is this: the review analyzed in a single pool four drugs that failed to meet clinical endpoints (e.g., ‘bapineuzumab’), ‘aducanumab (withdrawn due to safety concerns),’ and currently approved ‘lecanemab’ and ‘donanemab.’The argument is that grouping drugs with fundamentally different levels of development maturity, regulatory status, and mechanisms of action with equal weighting is scientifically inappropriate.Furthermore, the review is criticized for indiscriminately combining exploratory early-stage clinical trials (Phase 2) with large-scale confirmatory trials (Phase 3). Small-scale Phase 2 data with low statistical power were treated on par with Phase 3 results involving thousands of participants, leading to a severe distortion of the overall effect estimate. As a result, positive signals from drugs like lecanemab or donanemab, whose efficacy was clearly demonstrated in individual clinical trials, were diluted by the vast amount of noise from drugs that had failed in the past.Ji-won Seo, planning secretary of the Korean Dementia Association (Dongguk University Ilsan Hospital), stated, “If drugs at different development stages and regulatory statuses are combined, results will inevitably be diluted by the majority of data from failed drugs. It is not scientific to group successful drugs, failed candidates, and even those whose approvals have been revoked into the same basket and conclude that ‘the efficacy of this class is unclear.’”Regulatory approvals are based on independent dataGlobal regulatory authorities worldwide evaluate new drugs based on the specific clinical data unique to each drug, rather than on a generalized impression of the entire class.For example, lecanemab has been approved in over 50 countries worldwide, including the US FDA (July 2023), Japan PMDA (September 2023), China NMPA (January 2024), Korea MFDS (May 2024), UK MHRA (August 2024), and the European Commission (April 2025).Donanemab has also received sequential marketing authorization in over 40 major countries worldwide, including the US (July 2024), Japan (September 2024), China (December 2024), and Europe (September 2025).This global wave of approvals was made possible by thorough and independent reviews by regulatory agencies in each country, which evaluated the clinical evidence supporting these drugs’ ability to inhibit cognitive and functional decline, as well as the manageability of side effects such as ARIA. This means that, unlike the results of the Cochrane Review’s overly broad meta-analysis, their actual clinical value has already been validated worldwide.The Cochrane review cited ARIA as a major safety concern. ARIA is divided into cerebral edema (ARIA-E) and microbleeds (ARIA-H). While these are side effects that require careful management in the ATT class, the majority are asymptomatic or mild and can be adequately managed through regular MRI monitoring.These safety profiles have already been thoroughly reviewed during regulatory approval processes and are reflected in labeling and prescribing information.Alzheimer’s disease is ultimately a fatal, progressive neurodegenerative disorder. Now that evidence has been established that intervention during the early symptomatic stage can slow disease progression, unfounded misconceptions can lead to negative outcomes, causing patients to miss the “golden window” for treatment.Seo added, “With new treatments emerging in a field that previously had limited options, expectations for early diagnosis and treatment are increasing. However, following recent reports of research findings that were misinterpreted, many patients are expressing anxiety in the clinic or experiencing confusion when deciding on a treatment plan.”Seo emphasized, “We must remember that each drug has been approved by regulatory authorities based on clear scientific evidence. If the optimal window for treatment is missed due to misunderstandings, it could later result in far greater burdens for patients and their families.”

- Company
- "Data analysis of 500,000 patients…clinical utility of choline alfoserate reconfirmed"
- by Kim, Jin-Gu May 07, 2026 10:29am
- 'Choline alfoserate expert forum' recently hosted by DailyPharmA large-scale real-world data (RWD) analysis of 500,000 Korean patients with mild cognitive impairment (MCI) has reconfirmed the clinical utility of choline alfoserate in reducing the risk of progression to dementia.Experts evaluate that this study significantly strengthens the evidence base for prescribing choline alfoserate. It is considered academically meaningful because it revalidated the efficacy evidence from existing randomized controlled trials (RCTs) using massive datasets from actual clinical settings.Analysis of 500,000 Korean MCI patients...“Reduction in risks for both Alzheimer’s disease‧vascular dementia”At the 'Choline alfoserate expert forum' recently hosted by DailyPharm, Professors Han-kyeol Kim (Wonju Severance Christian Hospital), Choi Hojin (Hanyang University Guri Hospital), Lee Chan Nyung (Korea University Anam Hospital), and Kim Geon Ha (Ewha Womans University Mokdong Hospital), all neurologists, focused on the results of the study titled 'Effect of Choline Alfoserate: NHIS Cohort Study,' published last year.Professor Han-kyeol Kim (Wonju Severance Christian Hospital)Professor Han-kyeol Kim, who delivered the keynote presentation, introduced the research findings based on the National Health Insurance Service (NHIS) big data, which followed 508,107 patients newly diagnosed with MCI between 2013 and 2016.The study results showed that the choline alfoserate user group had a significantly lower risk of progression to Alzheimer’s dementia by 10.1% (HR 0.899) and vascular dementia by 16.8% (HR 0.832) compared to the non-user group. Furthermore, the risk of ischemic stroke decreased by 16.7% (HR 0.833), and the risk of hemorrhagic stroke decreased by 15.3% (HR 0.847).Professor Han-kyeol Kim said, "This was not a simple statistics of prescriptions. We linked health examination data to precisely adjust for variables such as smoking, alcohol consumption, income, and chronic diseases (hypertension, diabetes, dyslipidemia), as well as the duration of drug exposure to enhance objectivity," and added, "The study confirms that the use of choline alfoserate can be effective in reducing the risk of dementia progression and stroke in MCI patients, serving as a useful option for early intervention."Combining the scientific achievement of RCTs with the confirmed RWD… “Completing complementary evidence for dementia suppression”A discussion continued, chaired by Professor Choi Hojin, with Professors Lee Chan Nyung and Kim Geon Ha in attendance. They noted that this RWD study addresses the structural limitations of previous RCTs.The previous 'ASCOMALVA' study was designed as a multicenter, randomized, double-blind, controlled trial (RCT). The study targeted 210 patients with Alzheimer’s disease dementia accompanied by ischemic stroke, comparing a donepezil monotherapy group with a choline alfoserate + donepezil combination therapy group. The results confirmed significant improvements in the combination group across ▲the Mini-Mental State Examination (MMSE) ▲ Alzheimer's Disease Assessment Scale-Cognitive Subscale (ADAS-cog) ▲Instrumental Activities of Daily Living (IADL). Long-term follow-up over four years also showed that the rate of cognitive decline was inhibited in the combination therapy group.Experts agreed that this RWD study supplements the limitations of the RCT. Professor Choi Hojin evaluated, "The evidence for the efficacy of choline alfoserate has become even more robust by adding the analysis results of a large-scale Korean patient population to the ASCOMALVA findings," and added, "Results tracking the effects of drugs prescribed in the real world over a long period are highly meaningful as evidence from actual clinical practice, distinct from RCTs conducted in controlled environments."Professors Choi Hojin (Hanyang University Guri Hospital) ·Lee Chan Nyung (Korea University Anam Hospital) ·Kim Geon Ha (Ewha Womans University Mokdong Hospital)Professor Lee Chan Nyung stated, "While RCTs are suitable for proving drug efficacy in standardized environments, it is not easy to confirm actual conversion rates in slowly progressing diseases like dementia due to limitations in sample size and follow-up duration," and "This RWD study serves as powerful complementary evidence to existing RCTs by reaffirming the outcome of 'inhibiting dementia progression' through data accumulated over several years in actual clinical settings."“MFDS policy to enhance RWD reliability…increasing potential for utilization in clinical practice”Experts also emphasized that this study aligns with the recent government stance on the importance of RWD. In this regard, the Ministry of Food and Drug Safety (MFDS) announced the 'Guidelines for the Utilization of RWD/RWE in the Approval and Licensing of Medicines, etc.' in June 2024. Through this, the ministry presented specific criteria for actively recognizing RWD as clinical evidence for indication expansion or efficacy validation. In contrast, it had previously been used only as a supplementary tool for post-marketing surveillance (PMS) of side effects.The view among experts is that RWD research, which precisely analyzes NHIS big data from over 500,000 citizens, can yield new clinical evidence amid these changing regulatory environments. Professor Lee Chan Nyung emphasized, "As the vast RWD figures of 500,000 show, the dementia suppression effect confirmed in a large-scale patient group will serve as crucial evidence that gives clinical specialists the confidence to prescribe."Professor Kim Geon Ha said, "The 16.8% lower risk of progression to vascular dementia shown in this study holds great significance for the Korean elderly population with high vascular risk factors such as hypertension," and Kim concluded, "It raises the level of evidence for preemptive treatment of MCI patients in actual clinical settings."Professor Choi Hojin highlighted the public health significance of early intervention at the MCI stage. Choi added, "Delaying the onset of dementia by even just a few years can drastically reduce national medical costs and the caregiving burden on patients' families. Preemptive response through proven options will be a practical solution to alleviate the dementia burden in an aging society."
- Company
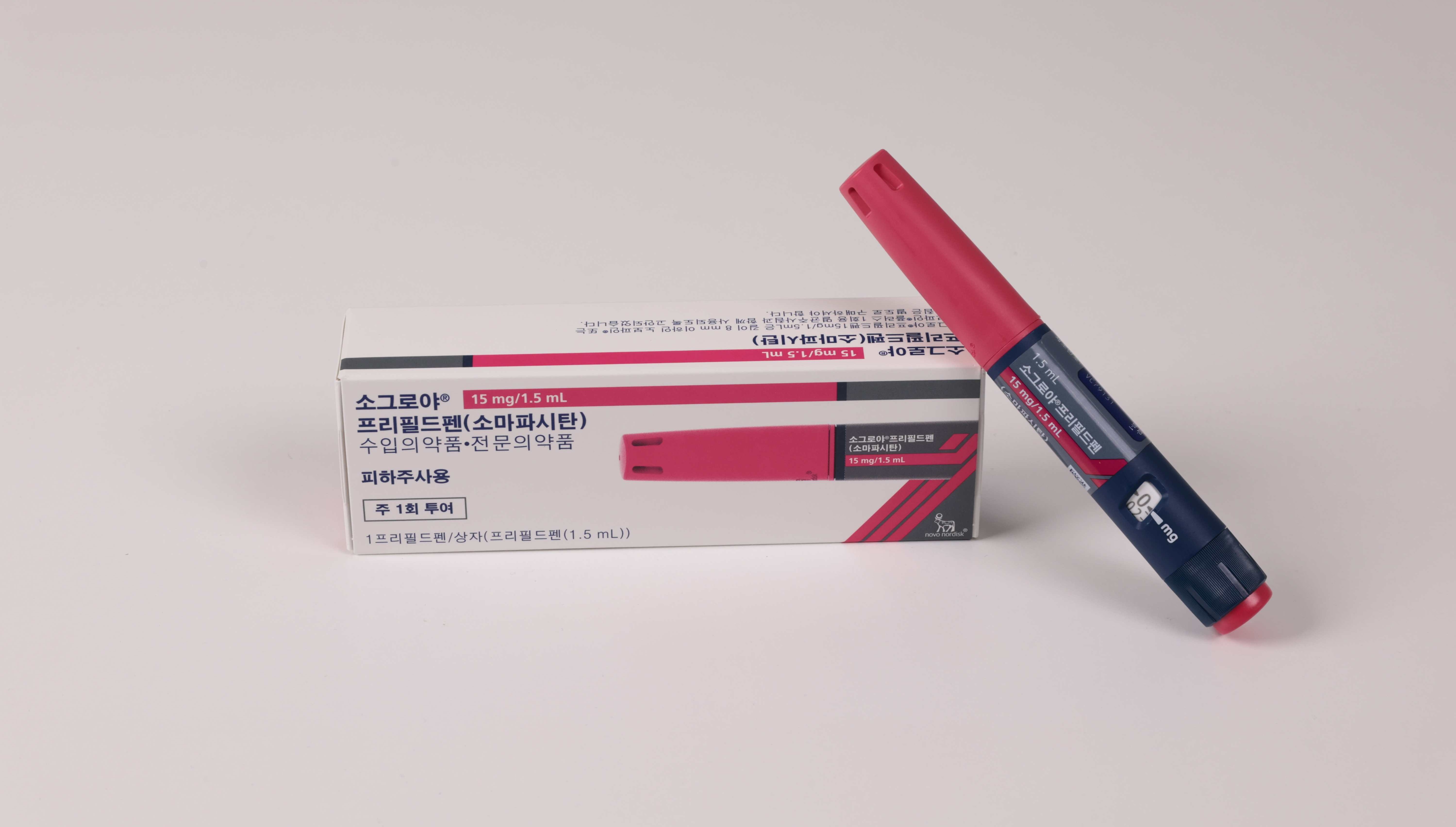
- Growth hormone deficiency drug Sogroya gains reimb in KOR
- by Son, Hyung Min May 07, 2026 10:29am
- Novo Nordisk Korea (General Manager: Kasper Roseeuw Poulsen) announced that starting on the 1st of this month, its long-acting, once-weekly growth hormone deficiency treatment ‘Sogroya Prefilled Pen (somapacitan)’ has been granted reimbursement for patients with growth hormone deficiency.Under the new reimbursement criteria, pediatric patients with growth hormone deficiency are eligible for Sogroya if they meet the following conditions: ▲ height below the 3rd percentile for chronological age, ▲ confirmed diagnosis through at least two growth hormone stimulation tests, ▲ and delayed bone age relative to chronological age.The recommended dosage is 0.16 mg/kg per week. Treatment is administered from a chronological age of 3 years until epiphyseal closure. However, reimbursement is limited to patients whose bone age falls within the range of 14–15 years for females and 15–16 years for males. However, patients within this category whose current height exceeds 153 cm (female) or 165 cm (male) must bear the full cost. In addition to children, Sogroya is covered for adult patients with growth hormone deficiency, provided certain criteria are met.Growth hormone deficiency is a condition characterized by delayed growth and may be accompanied by deficiencies in other pituitary hormones. Since consistent treatment is required until the end of the growth period, treatment adherence plays a critical role in treatment outcomes.The clinical efficacy and safety of Sogroya were confirmed in the global Phase III REAL4 trial.The REAL4 study was a randomized, parallel-group, open-label, active-controlled Phase III trial involving 200 treatment-naïve prepubertal pediatric patients with growth hormone deficiency. It evaluated the efficacy and safety of once-weekly Sogroya compared to once-daily growth hormone therapy.The primary endpoint was annualized height velocity (HV; cm/year) at Week 52. Secondary endpoints included changes in height velocity SDS, height SDS, the ratio of bone age (BA) to chronological age (CA), and IGF-1 SDS from baseline to Week 52.In the REAL4 study, Sogroya demonstrated non-inferiority in annual height velocity compared to daily growth hormone. At Week 52, the annual height velocity was 11.2 cm/year in the Sogroya group and 11.7 cm/year in the daily growth hormone group, showing comparable results.In terms of safety, the two treatment groups showed generally similar profiles. Most adverse reactions were mild or moderate in severity, and injection site reactions were reported in 5.3% and 5.9% of the Sogroya and daily growth hormone groups, respectively.Kasper Roseeuw Poulsen, General Manager of Novo Nordisk Korea, stated, “The reimbursement of Sogroya marks an important turning point in improving treatment access for patients with growth hormone deficiency. We expect that the once-weekly dosing option will improve treatment adherence, reduce the burden on patients and caregivers, and contribute to treatment continuity and improved quality of life.”

- Company
- Thyroid eye disease medicine 'Tepezza' lands in Korea
- by Son, Hyung Min May 07, 2026 10:28am
- Targeted therapy for thyroid eye disease (TED) 'Tepezza'Changes in treatment strategies are expected as the first targeted therapy for thyroid eye disease (TED) enters the Korean market.With follow-up candidates still in development or facing clinical hurdles with certain mechanisms, the possibility has been suggested that the market will remain centered on Amgen's already-commercialized Tepezza (teprotumumab).According to industry sources on the 7th, Amgen Korea recently obtained marketing authorization from the Ministry of Food and Drug Safety (MFDS) for Tepezza. The approved indication is for the treatment of adult patients with moderate-to-severe TED.TED is a rare disease in which autoimmune reactions cause inflammation and swelling of the orbital tissues. It causes proptosis, double vision, eye pain, and vision loss. If the disease progresses to a severe stage, it can lead to permanent disfigurement or even the risk of blindness.Until now, treatment in Korea has centered on symptomatic therapies such as steroids, radiation therapy, and orbital decompression surgery. However, critics have consistently pointed out that these have limitations in restoring structural changes such as proptosis or double vision.Tepezza is a monoclonal antibody that targets the Insulin-like Growth Factor-1 Receptor (IGF-1R), a key pathogenic mechanism in TED. It differentiates itself from existing treatments by targeting the disease to suppress the inflammatory response and tissue expansion.In the global Phase 3 OPTIC study, the proptosis response rate (a reduction of 2 mm or more) at 24 weeks was 83% for the Tepezza group and 10% for the placebo group. The diplopia (double vision) improvement rate also recorded 68% and 29%, respectively.In the OPTIC-J study of Japanese patients, the proptosis response rate was 89% in the Tepezza group and 11% in the placebo group.Tepezza already obtained regulatory agency authorization in the U.S. in 2020 and is currently the only approved targeted therapy for TED.FcRn inhibitors face hurdles… Delayed follow-up competitionSubcutaneous (SC) formulation 'Vyvgart (efgartigimod)'The competitive landscape for TED-targeted therapies remains limited, even in the global market.In particular, new drug candidates in the FcRn inhibitor class, which targeted to enter the TED market by reducing autoantibodies, are facing successive clinical barriers.Inhibition of the neonatal Fc receptor (FcRn), the protection receptor for Immunoglobulin G (IgG), is an approach that blocks the recycling pathway of IgG antibodies, thereby lowering the levels of pathogenic antibodies. It is evaluated as a treatment strategy applicable to autoimmune diseases in general. However, it appears to be struggling in the field of TED, beyond myasthenia gravis.HanAll Biopharma’s global partner, Immunovant, recently announced that its FcRn inhibitor batoclimab (IMVT-1401) failed to meet its primary endpoint in a global Phase 3 trial for TED. It failed to secure statistical significance in improving the Proptosis Responder Rate.Belgium-based Argenx also discontinued its clinical research on the FcRn inhibitor 'Vyvgart (efgartigimod)' for TED. Development was halted as the Independent Data Monitoring Committee (IDMC) judged the possibility of meeting the primary endpoints to be low.Accordingly, expert analysis suggests that a simple approach of reducing autoantibodies alone may be difficult to achieve sufficient clinical effects in TED.Chase by Viridian and Roche…the competition continuesNew drug development has not stagnated entirely. Among the followers, the most advanced is U.S.-based Viridian Therapeutics. Viridian is developing 'elegrobart,' a candidate based on the same IGF-1R mechanism as Tepezza.Elegrobart recently confirmed improvements in proptosis and diplopia in a global Phase 3 trial for patients with chronic TED, demonstrating the fastest rate of improvement among follow-up candidates.Roche’s 'Enspryng' However, Amgen has also moved to defend its market, recently reporting positive results from a Phase 3 trial of a subcutaneous (SC) formulation of Tepezza using an on-body injector (OBI). Consequently, competition in the TED market appears to be shifting toward competing for both efficacy and convenience, rather than simply SC conversion.Competition based on other mechanisms is also continuing. Roche’s 'Enspryng (satralizumab)' is a treatment that blocks the Interleukin-6 (IL-6) receptor, which is involved in inflammatory responses.Enspryng differs from the IGF-1R class, which directly targets ocular tissue expansion, by targeting the disease by suppressing orbital tissue inflammation and immune responses in TED.Roche recently announced positive results from the global Phase 3 SatraGO program for TED and disclosed plans to apply for authorization within the year.Enspryng failed to achieve statistical significance for the primary endpoint in the global Phase 3 SatraGO-1 study in patients with active TED. However, a pooled analysis with the follow-up Phase 3 study, SatraGO-2, reportedly confirmed a proptosis improvement effect.Until now, the treatment with the clearest clinical efficacy in TED has been the IGF-1R-targeted therapy. As FcRn inhibitors face successive clinical struggles, follow-up competition is trending toward a reorganization around IL-6R inhibitors and next-generation IGF-1R agents.
- Company
- A year after Leclaza securing European approval…Yuhan
- by Chon, Seung-Hyun May 06, 2026 03:28pm
- Yuhan expects to receive a $30 million technology fee payment for the European market entry of its new anticancer drug, Leclaza, soon.During the recent Q1 business performance presentation, Yuhan stated, “We expect to receive the $30 million milestone associated with the European launch of the lazertinib combination therapy soon,” and added, “The business plan established early this year is progressing smoothly.”Lazertinib, the active pharmaceutical ingredient of Leclaza, is a new anticancer drug developed by Yuhan. Leclaza is a non-small cell lung cancer (NSCLC) treatment that was approved in January 2021 as the 31st domestically developed new drug in Korea.In December 2024, the European Commission (EC) approved the combination therapy of Leclaza + Rybrevant as a first-line treatment for adult patients with locally advanced or metastatic NSCLC harboring epidermal growth factor receptor (EGFR) exon 19 deletions or exon 21 L858R substitution mutations.While the $30 million (approx. KRW 44 billion) milestone was met with Leclaza's entry into the European market, the payment has not yet been received, despite 17 months having passed. According to the company, the milestone will be transferred once sales of Leclaza begin in major European countries. Since the beginning of this year, Leclaza has been listed for health insurance in countries such as the UK, Switzerland, Italy, and Germany, and sales have begun. Securities analysts anticipate the European technology fee will be received within the first half of the year.Yuhan recorded KRW 5 billion in technology fee revenue in the first quarter. This is a 23.7% increase from the KRW 4 billion recorded in Q1 of last year, but significantly lower than the KRW 70.3 billion recorded in the previous quarter. Technology fee revenue inherently fluctuates, as it is generated from new drug licensing agreements or the advancement of development stages for licensed-out drugs.Last year, Yuhan received KRW 104.1 billion in technology fee revenue, with milestones from approvals in Japan and China accounting for a large portion.In May of last year, as the Japanese Ministry of Health, Labor and Welfare approved the Leclaza + amivantamab combination therapy, the requirement for an additional $15 million milestone was met, resulting in KRW 25 billion in technology fee revenue in Q2 of that year.In Q4 of last year, KRW 70.3 billion in technology fees were received. In August last year, China's National Medical Products Administration (NMPA) approved Leclaza as a combination therapy with Rybrevant, and Yuhan received a $45 million (KRW 69 billion) milestone from its partner, Janssen Biotech, for achieving the step-by-step milestone for Leclaza.Yuhan's Q1 technology fee revenue includes royalties from Janssen's sales of Leclaza. In 2024, the Leclaza and Rybrevant combination therapy received U.S. FDA approval, and sales in the U.S. began. According to Johnson & Johnson's performance data, Q1 sales of the Leclaza-Rybrevant combination therapy reached $257 million (approx. KRW 300 billion), an 82.7% increase year-on-year.Yuhan stated, “Prescription of the Leclaza combination therapy is expanding in major markets such as the U.S. and Europe, and we have secured a foundation for full-scale revenue growth through its listing as a 'Preferred Treatment' in the NCCN guidelines and the approval of the Rybrevant SC formulation.”In the 2026 National Comprehensive Cancer Network (NCCN) guidelines for NSCLC, the Leclaza + Rybrevant combination therapy was included as a 'Preferred Treatment' for first-line treatment. Leclaza is the first domestically developed new drug to be incorporated into the NCCN Category 1 first-line treatment.Yuhan received a steady inflow of technology fee revenue since 2018, when it began licensing new drug technologies.In July 2018, Yuhan licensed the technology for the degenerative disc disease treatment YH14618 to Spine BioPharma in the U.S. It received an upfront payment of $650,000 and was guaranteed $217.5 million in step-by-step milestones based on development, approval, and sales.In November 2018, Yuhan licensed out the anticancer drug Leclaza to Janssen Biotech. The total contract size, including a non-refundable $50 million upfront payment, is up to $1.205 billion.In January 2019, Yuhan signed a license and co-development agreement with Gilead Sciences for a new drug candidate targeting two drug targets for the treatment of metabolic dysfunction-associated steatohepatitis (MASH). The terms included a $15 million upfront payment and $777 million in milestones based on development, approval, and sales.In July 2019, Yuhan signed a technology transfer agreement for YH25724 with Boehringer Ingelheim. YH25724 is a dual-agonist targeting the GLP-1 protein and FGF21 factor simultaneously, with the technology transfer agreement signed during the preclinical stage. For this contract, Yuhan received a non-refundable upfront payment of $40 million. An additional $10 million milestone was generated for YH25724 in November 2021 upon entering Phase 1 clinical trials.In August 2020, Yuhan signed a technology transfer agreement with Processa Pharmaceuticals in the U.S. for YH12852, a treatment candidate for functional gastrointestinal disorders, in which Yuhan received a non-refundable $2 million upfront payment in the form of stock. Yuhan recognized the upfront payments and milestones received from the other four companies excluding Processa, which paid in stock, in installments. Yuhan received total technology fee revenue in 2019 until the first quarter of this year is amounted to be KRW 465 billion. Of the Leclaza technology fee revenue secured by Yuhan, 40% is paid to the original developer, Oscotec. In 2016, Yuhan acquired the development rights for Leclaza at the preclinical stage from Oscotec and its subsidiary Genosco. The total contract size for that acquisition was KRW 1.5 billion.
- Company
- Why are generic firms rechallenging Precedex's patent?
- by Kim, Jin-Gu May 04, 2026 10:33am
- An invalidation trial has been filed against the premix formulation patent of Pfizer’s sedative ‘Precedex (dexmedetomidine).’ What is interesting is that generic drug manufacturers had already succeeded in circumventing this patent back in 2020. In this context, some interpret this move as the generic companies’ attempt to launch a premix formulation and glass vial product containing the same active ingredient.According to industry sources on the 1st, Ilsung IS recently filed an invalidation trial against Hospira regarding the Precedex premix formulation patent.Precedex, approved in June 2010, is used for ‘sedation of patients who are intubated early under intensive care and receiving mechanical ventilation.’ Pfizer acquired the product through its acquisition of Hospira in 2015. Subsequently, Pfizer Korea received additional approval for the premix formulation in 2017.There are two patents related to Precedex. The substance patent expired in January 2013, leaving the formulation patent, which is set to expire in June 2032, as the current target of generic challenges.Generics were launched sequentially following the expiration of the substance patent. Companies including Ilsung IS, Hanlim Pharm, Penmix, Kyongbo Pharmaceutical, Pharmbio Korea, Hana Pharm, and Jeil Pharmaceutical obtained approvals. However, due to the active premix formulation patent, products were released in ampoule form, requiring mixing the dexmedetomidine-containing drug into a basic intravenous solution.In 2020, JW Life Science and Dai Han Pharm attempted to circumvent the premix formulation patent. JW argued that its proprietary container did not fall within the patent scope of Pfizer’s Precedex and won both first and second trials. It subsequently launched premix generics with its partners Hanlim and Hana Pharm. However, these products used special plastic infusion containers rather than the original glass vial.What is notable this time is that Ilsung IS chose to file an invalidation trial rather than a circumvention trial, as JW had filed. Given that circumvention trials generally have higher chances of success at first instance, Ilsung appears to have chosen a more difficult path.This is interpreted as an attempt to secure not only the premix formulation but also the glass vial packaging format.The premix formulation patent describes a ‘ready-to-use liquid pharmaceutical composition containing dexmedetomidine or its pharmaceutically acceptable salt thereof, placed in a sealed glass container for parenteral administration.’ Since the patent explicitly specifies a “sealed glass container,” its circumvention allows the sale of premix formulations but not in glass vials, as doing so would risk patent infringement.Glass vials and plastic infusion bags each have clear advantages and disadvantages. Plastic IV bags have the advantage of a relatively lower risk of breakage. Their light weight, which facilitates transportation, is also cited as an advantage. However, compared to glass vials, there is a greater risk of drug molecule leaching or adsorption. Additionally, reduced stability during long-term storage is pointed out as a drawback.On the other hand, glass vials prevent the drug from reacting with the packaging material, allowing it to maintain its purity for a long time. They also remain stable during long-term distribution and storage. This is why glass vials are the standard for pharmaceuticals that require long-term stability, despite the higher risk of breakage.The domestic market size for dexmedetomidine-based sedatives is estimated at around KRW 19 billion annually. According to the Ministry of Food and Drug Safety, domestic production and import volumes increased from KRW 16 billion in 2022 to KRW 19 billion in 2023, but declined back to KRW 16 billion in 2024 due to reduced usage amid the healthcare crisis. Pfizer’s two products account for about 40% of the total supply as of 2024.

- Company
- ‘Zipalertinib’ targets the Exon 20 lung cancer mkt…presenting new trt option
- by Son, Hyung Min May 04, 2026 10:33am
- While treatment options for EGFR exon 20 insertion mutation non-small cell lung cancer (NSCLC) remain limited, the potential for a change in the therapeutic environment is being proposed as the oral targeted therapy ‘zipalertinib’ enters the U.S. regulatory approval process.With consistent efficacy confirmed in global clinical trials and analysis of Asian patient populations amid limited existing treatments, zipalertinib is garnering attention as a competing product to ‘Rybrevant (amivantamab),’ the only approved medicine in this space.According to industry sources on the 4th, the U.S. Food and Drug Administration (FDA) recently accepted a New Drug Application (NDA) for zipalertinib. The drug’s target population includes patients with locally advanced or metastatic NSCLC with EGFR exon 20 insertion mutations whose disease has progressed following platinum-based chemotherapy. The goal review date under the Prescription Drug User Fee Act (PDUFA) is February 27, 2027.Zipalertinib is a next-generation irreversible EGFR tyrosine kinase inhibitor (TKI) being co-developed by Taiho Pharmaceutical of Japan and Cullinan Therapeutics of the U.S. This drug is designed to selectively inhibit mutant EGFR while minimizing impact on wild-type (normal) EGFR.The NDA is based on results from Part 2b of the Phase 1/2 REZILIENT1 study.The REZILIENT1 study included 176 patients with locally advanced or metastatic NSCLC harboring EGFR exon 20 insertion mutations as the primary efficacy evaluation group. The study evaluated the clinical efficacy of zipalertinib in patients whose disease progressed after platinum-based chemotherapy.Notably, 51 of the total patients had prior experience with Rybrevant, confirming zipalertinib’s potential as a subsequent treatment option after existing targeted therapies. Patients received 100 mg of zipalertinib orally twice daily, with objective response rate (ORR) and duration of response (DOR) analyzed as primary endpoints.Clinical results showed an ORR of 35.2% in the Zipalertinib group, with a median DOR of 8.8 months. It is noteworthy that responses were also confirmed in patients with prior Rybrevant treatment. In that specific patient group, the ORR was 30%, suggesting its viability as a follow-up treatment option.Zipalertinib results were presented at ESMO Asia 2025, held in Singapore last year.The efficacy in Asian patients did not differ significantly from that in the global patients.According to a subgroup analysis presented at the ESMO Asia 2025, the ORR for the Asian patient group was 33%, similar to the 37% observed in the non-Asian group. The duration of response (DOR) was 8.3 months and 10.5 months, respectively, while progression-free survival (PFS) showed almost identical patterns at 9.5 months and 9.0 months.In terms of overall survival (OS), the median has not yet been reached in the Asian patient group, whereas it was 24 months in the non-Asian group.Regarding safety, the primary adverse events included paronychia, rash, dry skin, diarrhea, and stomatitis, most of which were Grades 1–2 and manageable.Limited treatment environment…”Oral drug option is highly significant”Janssen’s RybrevantThe development of treatments for NSCLC with an EGFR exon 20 insertion mutation has faced significant challenges. Unlike treatments targeting Exon 19 deletions or Exon 21 L858R mutations, Exon 20 insertion mutations have diverse subtypes, making drug design structurally difficult.Takeda’s Exkivity, which garnered high expectations as an oral targeted therapy, received conditional approval based on a 28% ORR in early trials. However, it was withdrawn from the global market after failing to prove PFS improvement in the confirmatory Phase 3 trial (EXCLAIM-2).Poziotinib, previously developed, was also suspended after failing to meet efficacy expectations and experiencing toxicity issues in Phase 2 trials.As a result, Janssen’s Rybrevant is now the only treatment approved in this therapeutic area. However, as an intravenous-based therapy, it has been noted for limitations regarding administration convenience and treatment persistence.Given such a treatment gap, zipalertinib, which can be administered orally, is being evaluated as a new alternative because it offers manageable safety while increasing mutation selectivity compared to previous agents. The industry is focusing on the possibility that the Exon 20 mutation treatment landscape will transition from a monotherapy-centered environment to a more competitive one, depending on future approvals.Professor Ross Soo of the National Cancer Centre Singapore explained, “Zipalertinib demonstrated efficacy in Asian patients equivalent to that of the global patient population,” and added, “Because it is an oral drug, it is significant in terms of patient accessibility and treatment persistence.”

- Company
- Biosimilars expand access to osteoporosis treatment
- by Choi Da Eun Apr 30, 2026 08:24am
- The paradigm for osteoporosis treatment is shifting. There is a growing recognition that the condition should be approached as a ‘long-term management disease’ aimed at preventing fractures and maintaining quality of life, rather than merely slowing the decline in bone density. In particular, with the recent emergence of denosumab biosimilars improving treatment access, changes are being detected in the market as well.Professor Yoo Mee Kim of the Department of Endocrinology and Metabolism at Catholic Kwandong University International St. Mary’s Hospital emphasized, “Osteoporosis has almost no symptoms, so it is often first detected as a fracture before patients even realize they have the condition. Prevention and early treatment before fractures occur are most important.”Professor Kim has been serving as president of the Korean Society for Bone and Mineral Research since January 2026. She graduated from Yonsei University College of Medicine and earned her master’s and doctoral degrees from the same university. She currently heads the Endocrinology and Diabetes Center at International St. Mary’s Hospital. She is an active member of the Korean Endocrine Society, Korean Diabetes Association, Korean Society for the Study of Obesity, and other organizations.Professor Yoo Mee Kim. Director of the Endocrinology and Metabolism Center at International St. Mary’s HospitalOsteoporosis progresses without symptoms…fracture is the first signalAn osteoporotic fracture refers to a fracture that can occur even with a minor impact. It can even occur from a mild fall or coughing. If such a fracture occurs, it is necessary to suspect osteoporosis and undergo testing.Professor Kim explained, “Since osteoporosis causes no specific symptoms even as bones weaken, patients are often unaware of their condition. Consequently, many cases are only discovered after fractures occur in the wrist, spine, or hip.”The problem is what happens after a fracture. It does not simply end with a broken bone. She said, “When a fracture occurs, activity levels drop sharply and can lead to muscle loss and worsening chronic diseases. In elderly patients, reduced immunity may also increase the risk of complications such as pneumonia.”Hip fractures, in particular, are directly linked to survival. Professor Kim said, “When a hip fracture occurs in the elderly, it can lead to long-term hospitalization and complications, greatly increasing mortality. Even if a relatively minor fracture, such as a wrist fracture, occurs, it already means that the bones have weakened. Because fractures often begin at the wrist and progress to the spine and hip, early response is extremely important.”Osteoporosis increases sharply among women and after the 50s…menopause is a key variableOsteoporosis is particularly common in women. Professor Kim explained, “Female hormones play a role in protecting bones, but after menopause, hormone levels drop sharply, and bone loss progresses rapidly. From the 50s onward, prevalence and fracture risk increase significantly.”Recently, due to the aging population, the incidence of osteoporosis in men has also been on the rise. However, because hormonal decline occurs gradually in men, the onset of the disease tends to be slower than in women.Osteoporosis is diagnosed through Dual-energy X-ray absorptiometry (DXA). If a fracture has already occurred, or if there are factors such as advanced age, low body weight, family history, chronic disease, or polypharmacy, the patient is classified as high risk even if bone density is relatively good.Professor Kim emphasized, “Osteoporosis is diagnosed when the T-score, which indicates the degree of bone density loss compared with young adults, is -2.5 or lower. However, it is important to evaluate fracture risk alongside bone density measurements rather than relying solely on bone density values.”A shift in treatment paradigms… now capable of improving bone densityIn the past, osteoporosis treatment focused primarily on slowing progression. Today, major treatment options have diversified to include bisphosphonates, denosumab, and anabolic agents (PTH analogs, romosozumab).Bisphosphonates have long been the first-line treatment. Currently, denosumab-based formulations account for over 40% of the total osteoporosis treatment market share.Denosumab is characterized by a stronger effect through a mechanism that blocks bone resorption by inhibiting osteoclast formation. Since the emergence of denosumab, many changes have occurred in the osteoporosis treatment market.Professor Kim said, “While existing treatments were limited to slowing bone density loss, the recent emergence of more potent drugs has made it possible to actually increase bone density.”Daewoong Pharmaceutical’s osteoporosis treatment StobocloEmergence of biosimilars…raises expectations for improved treatment accessRecently, Prolia (denosumab) biosimilar ‘Stoboclo’ has been rapidly gaining traction in the market. It was first launched in March 2025. While the original drug exceeded KRW 200,000 when it first received reimbursement, Stoboclo was launched at about KRW 100,000, roughly half that level.When insurance reimbursement is applied, the actual cost borne by patients is only about KRW 180 per day and about KRW 5,400 per month on average. As a result, it is widely regarded as having significantly improved patient access. Just 10 months after its launch, it has established itself in the market with cumulative sales of KRW 11.8 billion.Professor Kim said, “If a drug has similar efficacy and safety, cost burden ultimately becomes an important factor from the patient’s perspective. Because osteoporosis treatment must be continued for a long period, a lower drug cost can have a positive impact on treatment persistence.”She continued, “Osteoporosis is not merely a disease of aging but a condition requiring active management. Preventing fractures through early diagnosis and continued treatment is of the utmost importance.”

- Company
- Outpatient Rx mkt growth slows amid reimbursement curbs and price cuts
- by Chon, Seung-Hyun Apr 30, 2026 08:24am
- The outpatient prescription market has shown steady performance this year. Analysts attribute this to a steady stream of flu patients and the expansion of insurance coverage for medications utilizing new technologies, such as oral anticancer drugs, which have spurred growth in the outpatient prescription market. However, the growth rate has slowed compared to previous years, partly due to reimbursement restrictions and drug price cuts.According to the pharmaceutical research institution UBIST on the 29th, the outpatient prescription market reached KRW 5.3484 trillion in Q1, a 3.5% increase year-on-year. This marks the third-largest quarterly market size on record, following Q3 and Q4 last year.Quarterly outpatient prescription volume (Unit: KRW 100 million, Source: UBIST)The outpatient prescription market has steadily expanded over time, growing 40.1% over five years from KRW 3.8173 trillion in Q1 2021.Following the COVID-19 endemic transition, persistent influenza and respiratory illness cases have sustained market growth. The Korea Disease Control and Prevention Agency reported that influenza alerts were issued continuously from January through last month, exceeding the epidemic threshold. For six months starting from Week 40 of last year (September 29–October 5), the incidence rate exceeded the seasonal epidemic threshold for the 2025–2026 season, 9.1 cases per 10,000 people.Proportion of Influenza-like Illness patient rates for the 2025–2026 season (Unit: Persons; Source: Korea Disease Control and Prevention Agency).Even without extraordinary factors like pandemics, the outpatient market is structurally set to grow due to aging populations and increasing chronic disease prevalence.Recently, expanded insurance coverage for innovative therapies has further driven market growth.For instance, AstraZeneca’s anticancer drug Tagrisso recorded KRW 54.2 billion in outpatient prescriptions in Q1, a 26.2% increase year-on-year. Tagrisso is used for EGFR-mutated metastatic non-small cell lung cancer (NSCLC). Since 2024, its reimbursement has expanded alongside Yuhan’s Leclaza for first-line treatment of certain mutation-positive NSCLC.Although anticancer drugs are typically used more in inpatient settings, Tagrisso’s oral formulation has significantly boosted outpatient prescriptions. Its outpatient sales more than doubled from KRW 22.7 billion in Q1 2023 to current levels. Leclaza also saw Q1 prescriptions reach KRW 19.1 billion, more than tripling in three years.However, Q1 growth appears to have slowed somewhat compared to previous years. This marks the lowest growth rate in five years, following a 2.9% year-over-year increase in prescription sales in Q1 of 2021. The outpatient prescription market grew by 10.9% year-over-year in Q1 2022 and expanded by 9.9% in Q1 2023. In Q1 2024 and last year, the market grew by 5.5% and 5.3% year-on-year, respectively. The strong upward trend continued as infectious disease patients continued to emerge amid the COVID-19 pandemic and endemic phase.Government drug pricing policies are cited as a major factor. Repeated reimbursement restrictions and price cuts have dampened market expansion.For example, the brain function enhancer choline alfoscerate market shrank from KRW 146.2 billion in Q1 last year to KRW 103 billion this year, a 29.5% drop in one year.Since September 21st, the patient co-insurance rate has increased from 30% to 80% for non-diagnosed patients, sharply reducing prescriptions. In Q4 last year, the prescription market for choline preparations stood at KRW 103.7 billion, a 29.9% decrease from the previous quarter’s KRW 147.9 billion, and remained at a similar level in Q1 this year. This means that over the past six months, the prescription market for choline preparations has shrunk by more than KRW 90 billion due to the reimbursement cut.In Q1, the outpatient prescription market for mugwort extract-based drugs stood at KRW 25.3 billion, a 18.7% decrease year-on-year. Mugwort extract-based drugs are natural medicines made from mugwort and are used to improve gastric mucosal lesions, bleeding, redness, and swelling associated with acute and chronic gastritis. Following the government’s reimbursement reevaluation, the insurance price ceiling for 74 types of mugwort extract-based medicines was reduced by an average of 14.3% starting in February, causing the prescription market to contract.Unexpected variables also impact the market. The COVID-19 drug Paxlovid recorded KRW 47.7 billion in prescription sales in Q3 last year, but this figure shrank to KRW 2.1 billion in Q1 this year. Paxlovid is primarily prescribed to high-risk patients at risk of severe progression. Initially, the government purchased and supplied it free of charge, but in June 2024, the government halted the supply of new batches, shifting to regular prescriptions through general medical institutions.With the decision on Paxlovid’s health insurance reimbursement in October 2024, it officially entered the prescription market. The reimbursement price was set at KRW 941,940, with a 5% co-insurance rate. Due to the fluctuating number of COVID-19 patients and Paxlovid’s high price, there was significant variation in prescription volume. Paxlovid’s prescription sales of KRW 47.7 billion in Q3 last year accounted for 0.9% of total outpatient prescription sales.
- Company
- Boryung loses first trial in Lenvima + Keytruda combo patent dispute
- by Kim, Jin-Gu Apr 30, 2026 08:24am
- Boryung has lost the first trial in a patent dispute over the combination therapy of the liver cancer treatment Lenvima (lenvatinib) and Keytruda (pembrolizumab). Although Boryung had already launched a generic after overcoming listed patents, the latest ruling has left the company facing marketing risks for key indications. To mitigate this risk, the company must now seek a reversal of the ruling by appealing to the Intellectual Property Court.According to industry sources on the 29th, the Intellectual Property Trial and Appeal Board recently issued a ruling of partial dismissal and partial rejection in a passive scope confirmation trial filed by Boryung against Eisai Korea and Merck regarding an unlisted patent (No. 10-2662228) related to the Lenvima + Keytruda combination therapy. The IPTAB also dismissed the invalidation trial request for the same patent.This patent covers the ‘combination of a PD-1 antagonist and a VEGFR/FGFR/RET tyrosine kinase inhibitor for treating cancer.’ Merck and Eisai registered this patent in April 2024. The patent covers the combination therapy of Lenvima and Keytruda for the first-line treatment of advanced endometrial cancer and advanced renal cell carcinoma.Among Lenvima’s indications - ▲ locally recurrent or metastatic progressive differentiated thyroid cancer, ▲first-line treatment for unresectable hepatocellular carcinoma, ▲combination therapy with pembrolizumab for the treatment of patients with advanced endometrial cancer, ▲combination therapy with pembrolizumab as first-line treatment for advanced renal cell carcinoma - this case is related to the 3rd and 4th indications.Industry analysis suggests that Boryung has effectively suffered a decisive defeat in the first-instance trial over the Lenvima + Keytruda combination patent. However, the ruling does not block the sale of Boryung’s Lenvima generic, Lenvanib. Previously, Boryung had bypassed or invalidated all four listed patents for Lenvima except the substance patent, and received marketing approval for Lenvanib in February last year. It then secured reimbursement listing in July and launched the product. Currently, Boryung’s Lenvanib is the only Lenvima generic on the market.However, due to this ruling, Lenvanib can no longer be used to treat advanced endometrial cancer and advanced renal cell carcinoma. This is because if Lenvanib is used for these indications, Boryung could face patent infringement lawsuits from Eisai and Merck.Eisai has already demonstrated a proactive stance in protecting its patent rights, as evidenced by its filing of an active scope confirmation trial regarding the scope of rights during a dispute over another use patent (10-1470653) against Boryung. Although that case concluded with Eisai voluntarily withdrawing its claim following Boryung’s victory in the invalidation trial, had the invalidation trial yielded the opposite result, it could have served as grounds for a patent infringement lawsuit.At present, Lenvima’s core market remains thyroid cancer and hepatocellular carcinoma (indications 1 and 2). However, Boryung’s concerns have grown as Lenvima’s focus in the global market is rapidly shifting toward combination therapy with Keytruda for endometrial cancer and renal cell carcinoma.To resolve this patent risk, Boryung must now decide whether to appeal. Industry observers expect that, given the growing importance of combination therapy in Lenvima’s global revenue mix, Boryung is unlikely to accept the first-instance defeat without a challenge.