- LOGIN
- MemberShip
- 2026-05-07 18:32:28

- Policy
- Targets for PVA in third quarter include Darzalex & Alunbrig
- by Lee, Hye-Kyung Jul 01, 2021 05:55am
- Janssen Korea's (Daratumumab) and Takeda Korea’s Alunbrig (Brigatinib) were included in the Price-Volume Agreement (PVA). Otsuka Pharmaceutical Korea's "Iclusig" and AstraZeneca Korea's "Impinji" (Dubalumab) were also subject to negotiations on drug prices due to increased usage. The NHIS recently introduced "PVA (Type Ka/Na) Monitoring Drugs for 3rd Quarter of 2021" on its website. Targets for monitoring in third quarter of this year are 173 items from 90 pharmaceutical groups. PVA is a method shared by the NHIS and pharmaceutical companies to share the risk of health insurance finances, and the drug price will be reduced through negotiations with the NHIS. Type Ka of PVA corresponds to cases in which claims for the same product group with expected claims agreed with the NHIS have increased by more than 30% from expected claims. Type Na is subject to the same product group, which has been negotiated for type Ka or not negotiated for type Ka, where the previous type has increased by more than 60% or more than 10% or ₩5 billion every year from the end of the analysis period. Drugs with an annual claim of less than ₩1.5 billion, drugs with lower upper limit than the arithmetic average of the same ingredients, low-cost drugs, and drug shortage prevention programs are excluded from PVA. The pharmaceutical groups included Samoh's "Vimizim (Elosulfase Alpha)," Sanofi-Aventis Korea's "Mozobil (Plerixafor)," BL&H's Trisenox(Arsenic Trioxide) and SK Chemical's Migard(Frovatriptan Succinate Monohydrate). The pharmaceutical groups included Samo Pharmaceutical's "Vimizim (Elosulfase Alpha)," Sanofi-Aventis Korea's "Mozobil (Plerixapor)," BL&H's "Arsenic Trioxide" and SK Chemical's "Migard" (Frovat Monochlithinccate).

- Policy
- 2 indications deleted for choline alfoscerate, changes label
- by Lee, Tak-Sun Jun 30, 2021 05:56am
- Authorities have ordered companies to delete the No.2 and No.3 indication of the brain function enhancer choline alfoscerate, for which the clinical re-evaluations have started recently. Accordingly, the indication must be removed from the label from the 28th of next month. Also, the Ministry of Food and Drug Safety requested doctors and pharmacists to review the need to prescribe and dispense substitute drugs for patients that use choline alfoscerate during the labeling change period. On the 28th, the MFDS ordered a label change for 144 choline alfoscerate products. After a review of the clinical trial protocols for the reevaluation of these products, the ministry determined that that trial will be unable to assess the efficacy of choline alfoscerate’s second and third indications and decided to remove the two indications. As a result, only the first and main indication, for ‘Secondary symptom caused by cerebrovascular defects or degenerative brain-organic psychiatric syndrome by cerebrovascular deficiency: reduced memory, derangement, disorientation by reduced willingness and spontaneity, reduced willingness and spontaneity, reduced concentration’ will be left approved for choline alfoscerate. The second indication, for ‘Emotional and behavioral change: emotional insecurity, sensitive to stimulation, indifferent to surrounding,’ and the third indication for’ Senile pseudodepression’ will be deleted. However, whether the first indicating will remain intact depends on the results of the domestic clinical reevaluation that will be conducted. The MFDS had ordered companies to submit clinical trial results for Alzheimer's patients by December 9th, 2025, and the same for patients with mild cognitive disorder by March 9th, 2025. 57 companies including Daewoong Pharmaceutical and Chong Kun Dang that will lead the clinical trial will be participating in the clinical reevaluation. With the order to change labels issued, choline alfoscerate products that will be produced a month from the indication change, from July 28th, must reflect the new efficacy and effect on the product box and label. Administrative sanctions will be imposed on the companies that do not comply with the order by the said date. In 2019, the prescription sales amount of choline alfoscerate for the deleted two indications (No.2, No.3) amounted to 39.3 billion won and accounted for 11.1% of total prescription sales for the drug. Therefore, pharmaceutical companies are not expected to suffer huge performance losses from the change of indications. The companies are more intently focused on negotiations with the government on the amount they may have to give back in case the reevaluations for the first indication fall through. The National Insurance Health Service had recently proposed a retrieval rate of 30% of the insurance claims amount to make progress in the fruitless negotiations.
- Policy
- Will Rosuvastatin OD be released?
- by Lee, Tak-Sun Jun 30, 2021 05:55am
- Crestor (Rosuvastatin)Attention is focusing on whether a treatment for hyperlipidemia, Rosuvastatin OD (Orally Disintegrated Tablet) will be released. Rosuvastatin OD was approved in Japan in 2016, but has not yet been released in Korea. According to industries on the 25th, BCWORLD Pharm recently applied for approval of Rosuvastatin OD to the MFDS. AstraZeneca's Crestor is the original for Rosuvastatin. Generics have been released in Korea since 2009, and 941 cases are currently approved. However, there is no formulation of ODT that is melted with the tongue without water. In Japan, AstraZeneca and Shionogi were approved for Crestor OD in 2016. It is said to have been developed by Shionogi. ODT is useful for patients who have difficulty swallowing tablets or who need to refrain from drinking water. However, since most patients are familiar with regular tablets, there will not be much demand for ODT formulation. Last year, Crestor recorded ₩85.5 billion in outpatient prescriptions in the country. Therefore, if ODT is released for the first time, it is enough to attract attention regardless of market size. However, it is too early to discuss the success of commercialization. When a new drug is applied for permission, the MFDS decides whether to approve the sale after safety and the review.
- Policy
- Janssen Vaccine for 10,800 people has been approved
- by Lee, Tak-Sun Jun 29, 2021 05:46am
- The MFDS said it approved the nation lot release on the 25th (Friday) for 108,800 people of "Covid-19 Vaccine Janssen" applied by Janssen Korea. Nation lot release is a system in which the state checks the quality of the vaccine once again before it is distributed on the market by comprehensively evaluating the MFDS' test results of the vaccine. The MFDS explained that it has been thoroughly preparing for rapid national lot release of "Covid-19 Vaccine Janssen" by verifying its own test method since early this year and introducing equipment needed for testing such as enzyme analyzers. Test methods established by the MFDS include implantable gene expression, viral gene verification, viral protein verification, vector content, and purity. The effectiveness, safety, and quality were confirmed through the test and manufacturing and test data review of Covid-19 Vacine Janssen Inj and the lot release was decided because it met the national shipping approval criteria. This product was tested for the effectiveness of vaccines such as ▲ antigen protein expression, viral gene confirmation indicating effectiveness, and viral gene and transporter (vector) content by conducting a ▲ antigen test, verification test, etc. ▲It was confirmed that the product was not contaminated by conducting aseptic test, endotoxin test, and purity test. ▲ The MFDS explained that quality consistency was confirmed by conducting quality, pH, and practicality tests and reviewing quality test data issued by the manufacturer's quality assurance manager. Covid-19 Vaccine Janssen Inj is a virus vector vaccine manufactured by incorporating COVID-19 surface antigen gene into the adenovirus mold, the same platform as the country's first licensed AstraZeneca Korea Covid-19 Vaccine Inj. The MFDS stressed that it will do its best to thoroughly verify COVID, which will be introduced in Korea, by making the most of the related infrastructure necessary for the national lot release. Janssen vaccine, which was approved for lot release this time, is not a vaccine donated by the U.S. government, but an individual contract with Janssen.
- Policy
- Leclaza was listed at ₩68,964 per tablet
- by Kim, Jung-Ju Jun 29, 2021 05:46am
- Leclaza 80mg (Lazertinib) made with domestic technology passed the final step to insurance benefits. Leclaza finally chose this track, considering its similar clinical utility to the alternative drug Tagrisso, but cheaper with the Risk Sharing Agreement (RSA) Refund and Expenditure Cap. The date will be started on July 1, which means that the company succeeded six months after the approval in January. The MOHW announced on the 25th that the revised "Pharmaceuticla benefit list and upper price limit table" was proposed as a sub-issue of the 15th Health Insurance Policy Committee. Leclaza is a third-generation epithelial cell growth factor receptor (EGFR) tyrosynkina inhibitor (TKI) and received "conditional approval" from the MFDS on January 18 this year as a treatment for local progressive or metastatic non-small cell lung cancer. At that time, the company applied for insurance registration at the HIRA on December 30 last year, just before being licensed using the drug approval-patent linkage system. On 24 February, the HIRA held the Cancer Drugs Benefit Appraisal Committee to discuss the drug, and was judged on 8 April after deliberation by the Pharmaceutical Benefits Advisory Committee. The Pharmaceutical Benefits Advisory Committee determined that the clinical usefulness was similar to that of Tagrisso, an alternative drug, and that it was therapeutically equivalent to Tagrisso, which is currently being applied to RSA. Leclaza also concluded that following the RSA track would be cost effective at a lower cost than Tagrisso. Korean Cancer Study Group, Korean Society of Medical Oncology, and Korean Association for Lung Cancer suggested these opinions that similar effects have been shown compared to Tagrisso, and it is an additional treatment alternative that demonstrates safety results such as decreased cardiac toxicity and is more effective than treatment. It chose refund and expendature cap among RSA. The RSA Refund type is that a pharmaceutical company reimburses the NHIS for the full amount of pre-determined drug claims, and the Expenditure Cap reimburses the NHIS for a certain percentage of the excess if the actual claims exceed the pre-set annual expected cap. Since then, the MOHW has ordered negotiations on drug prices between the NHIS and the company and agreed on the estimated amount of claims for Refund and Expenditure Cap from April 23 to the 8th of this month. It has agreed that it will make $18.5 million (₩14.1 billion). The NHIS predicts no additional financial needs because there is an alternative drug, Tagrisso. Leclaza was insured from authorization in just half a year. The price is ₩68,964 per tablet, and the benefit will begin in earnest on the 1st of next month.
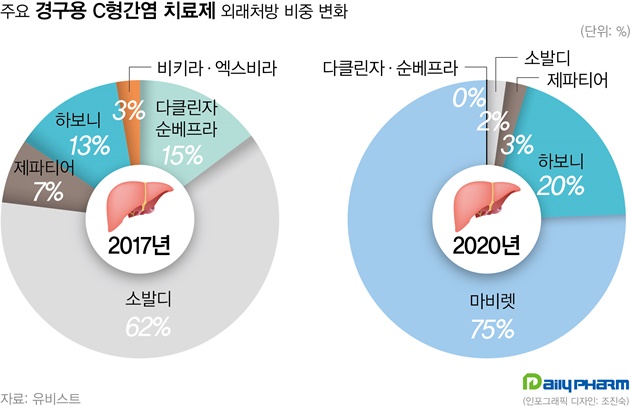
- Policy
- Following Gilead's hepatitis C tx Epclusa, Vosevi was filed
- by Lee, Tak-Sun Jun 28, 2021 05:50am
- This year, hepatitis C drugs Epclusa and Vosevi have been filed by the MFDS in Korea. These items are expected to replace Sovaldi. According to the industry on the 27th, Gilead's Vosevi (Sofosbuvir/Velpatasvir/Voxilaprevir) has recently been filed and will begin screening in earnest. Vosevi is expected to be a Pangenotypic DAA preparation that can be used in patients who fail to treat existing Direct-Acting Antiviral (DAA) drugs. AbbVie's Mavyret, a phangenotypic inhibitor, is now the market leading item. And, Mavyret is applicable to both chronic hepatitis C virus genotypes 1, 2, 3, 4, 5 and 6. For Gilead's Sovaldi, it is only valid for type 1, 2, 3 and 4 and must be administered simultaneously with Ribavirin. For this reason, Mavyret currently occupies 75% of the domestic hepatitis C treatment market based on UBIST last year. Sovaldi, by contrast, stood at 2%. Sovaldi had a 62% market share in 2017. Gilead's other hepatitis C drug, Harvoni, came in second with a 20% share after Mavyret last year. Epclusa (Velpatasvir + Sofosbuvir) was also received. Epclusa is also a pangenotypic treatment that can be administered regardless of genotype, a competitive drug for Mavyret. If the two drugs are registered in Korea, it is predicted that they will replace performance of Sovaldi and Harvoni. Although the market for hepatitis C drugs is decreasing in size due to the decrease in the number of patients due to effective drugs, it is attractive for pharmaceutical companies as Mavyret achieved ₩32.6 billion in outpatient prescription last year. As Gilead prepares a new drug, the domestic hepatitis C treatment market is expected to enter fierce competition again.
- Policy
- 16 drugs including Venclexta receive reimbursement in 1H
- by Kim, Jung-Ju Jun 28, 2021 05:50am
- A total of 14 products were newly listed on the insurance benefit list to improve patient access in the first half of this year. Also, two drugs benefited from the reimbursement criteria expansion that was applied to already-listed new drugs. This expanded coverage is interpreted as a result of the government’s decision to flexibly apply coverage for drugs according to the patients’ needs and social importance. The government estimates that around 760,000 patients in Korea will be benefiting from the change. The coverage will financially cost around 115.6 billion won per year, which indicates that the coverage expansion for drugs is being carried out faster than the previous year. During the first half of this year, from January to June, a total of 16 drugs (based on each drug’s representative strengths) were listed on the drug benefit list. These include new drugs that were newly listed and those whose criteria (indication, administration criteria, etc) were expanded to enhance coverage. New drugs that were newly listed this month include the hemophilia A treatment Afstlya inj.; bacteremia treatment Daptocin inj., Boryung Daptomycin inj., and Dapto Inj.; nocturnal hemoglobinuria treatment Ultomiris inj.; and hyperlipidemia treatment Praluent Pen inj.. Also, the reimbursement criteria for the already-listed choric lymphocytic leukemia treatment Venclexta tab. was expanded to enhance coverage. The number of domestic patients expected to use the drugs that were newly listed or received expanded reimbursement varies greatly by product. For the Venclexta tab., the number of patients that will benefit from the expanded reimbursement is expected to be around 75. Also, the number of patients to benefit from the newly listed Ultomiris is 92. Contrary to such drugs that are expected to benefit less than 100 patients, the glaucoma treatment Eybelis Eye Drops is expected to benefit around 45,000 patients. Also, the blood sugar regulator Xultophy Flex Touch inj. that was newly listed in May is expected to benefit 12,756 patients, and Parkinson treatment Equfina Film Coated tab. that started its reimbursement process in January this year is expected to benefit 7,000 patients. The number of beneficiaries greatly varies due to policies that now allow flexible expansion of NHI coverage to high-price drugs that are used for a small number of rare diseases, that were established based on the increased social maturity that can now accept such policies. Due to the new listings and expanded benefit criteria, the government and payer are expected to spend around 115.6 billion won a year to grant access to around 76,769 patients.
- Policy
- Billing for α-GPC is managed
- by Lee, Jeong-Hwan Jun 28, 2021 05:50am
- The HIRA plans to consider the need to manage claims by selecting Choline alfoscerate, which is under controversy over reducing adaptation certificates, as the "selection focused item." The MFDS plans to periodically monitor pharmaceutical companies that have been clinically reassessed, block unnecessary clinical extensions, and take administrative action against pharmaceutical companies that have failed to reassess. On the 24th, the HIRA and the MFDS responded to Nam In-soon's criticism of the Democratic Party of Korea about Choline alfoscerate. She ordered the HIRA to review and implement follow-up measures to prevent Choline alfoscerate from prescribing drugs based on reduced benefit due to the lack of proven efficacy and effectiveness of other than dementia. The HIRA said it will collect opinions from the public and medical circles and decide whether to select and manage Choline alfoscerate as a selective target after deliberation and resolution process of the central screening coordinator committee. It is a proactive screening system that selects items that need to improve medical trends, such as increased medical expenses, screening problems, and social issues, and makes intensive screening after prior notice to induce improvement in autonomous medical trends of medical institutions. The HIRA will make efforts to make sure that Choline alfoscerate is not prescribed for other than dementia in front-line medical institutions. In addition, the HIRA said it is currently monitoring claims, amounts, and actual number of employees after the suspension of execution of Choline alfoscerate's benefit standard notice. "We will continue to monitor Choline alfoscerate's claims and come up with follow-up measures with related agencies," The HIRA said. "We will continue to re-evaluate the benefit adequacy of the system to streamline spending structure for health insurance fiscal sustainability of health insurance." She ordered the MFDS to block pharmaceutical companies from indiscriminately extending the clinical re-evaluation period of Choline alfoscerate and start managing pharmaceutical companies that have not submitted clinical plans. The MFDS said it will monitor the progress of clinical re-evaluation periodically and speed up the process of changing or canceling permits if it is deemed ineffective or insufficient. The MFDS said 11 items from eight companies that did not participate in the clinical re-evaluation are being subject to secondary administrative measures under the Pharmaceous Affairs law. The first disposal will be suspended for two months, the second disposal will be suspended for six months, and the third disposal will be revoked. The MFDS announced on the 10th that it had previously notified the procedure of changing permission to delete "emotional and behavioral changes, senile caustic depression," which is not included in the scope of clinical re-evaluation. The intention is that there is a need for management only for "secondary symptoms and metamorphic or degenerative cerebral substrate syndrome caused by cerebrovascular deficits" where clinical re-evaluation has been decided. The MFDS said, "To reevaluate the validity quickly, we adjusted and approved the clinical trial period after consulting experts based on the progress of clinical trials of similar adaptive items and data on insurance claims by the HIRA." The MFDS replied, "We will closely review the re-evaluation process and take administrative action against non-submitted pharmaceutical companies."
- Policy
- The number of items has plummeted due to regulatory impact
- by Lee, Tak-Sun Jun 28, 2021 05:49am
- Recently, More and more items are being removed due to the expiration date. Pharmaceutical companies have sorted out the items due to strong regulations on impurities and penalties for drug prices of consignment items. According to the MFDS on the 23rd, a total of 63 new drugs have been approved this month. On the contrary, there are 226 items which permits have been deleted, almost four times more. From January 1 to June 23 this year, 1,224 items were licensed. On the other hand, 2,229 items were deleted, nearly doubling. Most of the deleted items have expired five-year period, and they have not submitted data that proves the safety and validity of the license renewal. Since 2018, the MFDS has operated a renewal system for item evaluation and has been evaluating it every five years. Pharmaceutical companies are required to submit data such as safety management, quality, and status of foreign use to renew their permits. As a result, items withdrawing from the market are often given up their permits. Analysts say that this pattern has become worse recently. An industry official said, "We submitted an evaluation of the possibility of impurities for all medicines by June, and decided not to renew the license because some items are difficult to proceed." An official from another company said, "Most of the consignment items are given permission as penalties are given at drug prices." "There have been far more cases of deletion recently because there are no items to put on the market," he said. Items subject to regulation due to the detection of impurities are likely to be removed. Satan or Tidin drugs, which have been a problem recently, are representative. Even if these items are sold, lot release is possible only when tests prove that there are no impurities.

- Policy
- NA LJC to review ‘1+3 bill for generics’ tomorrow
- by Lee, Jeong-Hwan Jun 25, 2021 05:51am
- The National Assembly's Legislation and Judiciary Committee will review the revision of the Pharmaceutical Affairs Act that contains the bill for the ‘1+3 restriction for bioequivalence and clinical test’ of generics and drugs requiring data submission an agenda for review at the NA Legislation and Judiciary Committee’s general meeting that will be held on the 25th. If the revision passes the Legislation and Judiciary Committee, it will be processed and deliberated at the plenary session that is planned on the 29th, and may likely be legislated by June. On the 24th, the vice-chairpersons of the ruling and opposition party of NA’s Legislation and Judiciary Committee are discussing the agendas that will be presented at the plenary session on Monday. One thing to note is that the bills passed by the Health and Welfare Committee on the 16th, including the one to revise the Pharmaceutical Affairs Act, have also been included as agendas at the Legislation and Judiciary Committee meeting. The revised Pharmaceutical Affairs Act passed by the Health and Welfare Committee includes the ‘1+3 restriction for bioequivalence and clinical test’ of generics and drugs requiring data submission, designation of Day of Medicine as a Statutory Anniversary, mandatory preparation and submission of Contract Sales Organization (CSO) expenditure report, penalizing illegal purchasers of specialty drugs, and compulsory marking of safety precautions in Braille and voice codes. The revision also includes the bills to legislate the conditional approval system for Phase III trials, mandatory registration of oversees API manufactories, increasing the number of Central Pharmaceutical Affairs Council members to 300 at most, strengthening regulations for the cancelation of false or unlawfully approved National Lot Release drugs, and the establishment of the Vaccine Safety Technology Support Center. In other words, whether the bill to regulate generics and drugs requiring data submission will be able to pass the final legislative threshold by passing the plenary session of the National Assembly this month will depend on deliberations of the Legislation and Judiciary Committee on the 25th. The Legislation and Judiciary Committee can decide to pass or continue to review the bills passed by the Health and Welfare Committee. If the bill is passed by the Legislation and Judiciary Committee, the final procedure will be carried out at the plenary session on the 29th. When the government finally promulgates the bill, the individual bills will then come into effect according to the by-laws. An official from the Legislation and Judiciary Committee said, “Discussions on the agenda for the plenary session have not yet been completed by the vice-chairpersons. For now, the bill by the Health and Welfare Committee, including the revised Pharmaceutical Affairs Act, is included in the agenda for the plenary session."