- LOGIN
- MemberShip
- 2026-07-22 06:54:19
- Policy
- Twice-yearly injectable HIV drug Sunlenca nears approval in Korea
- by Lee, Tak-Sun Mar 20, 2026 08:55am
- AI-generated imageGilead Sciences’ multidrug-resistant HIV treatment Sunlenca (lenacapavir sodium) is reportedly nearing marketing approval in Korea.The drug was designated in March last year for the MFDS GIFT (Global Innovative product on Fast Track) program, and it has recently been reported that the safety and efficacy review has been completed.According to industry sources on the 19th, the MFDS has completed its safety and efficacy assessment of Sunlenca. Once a product passes this stage, only the final approval process typically remains. In addition, products that have undergone safety and efficacy reviews can apply for reimbursement to the Health Insurance Review and Assessment Service (HIRA) under the approval–reimbursement assessment linkage system.Observers predict that if Sunlenca has received a positive determination in the safety and efficacy review, it is highly likely to receive marketing authorization approval in the near future.Sunlenca is a first-in-class capsid inhibitor that targets multiple stages of the HIV replication cycle. It is primarily aimed at patients with multidrug-resistant HIV who have developed resistance to existing treatments and have no further treatment options.It inhibits HIV-1 at multiple points throughout the viral lifecycle, including by interfering with the capsid-mediated nuclear entry of the pre-integration complex and impairing virion production and proper capsid core formation.A key feature of the drug is its twice-yearly injectable dosing, offering improved convenience.This is because it is a “long-acting” formulation that requires only a single injection every 6 months (26 weeks) during the maintenance phase, after an initial phase combined with oral therapy. As a result, it is widely recognized for significantly reducing the psychological burden and inconvenience previously experienced by patients who had to take medication daily.An industry official stated, “Sunlenca is of great clinical significance in that it provides strong viral suppression through a completely new mechanism for these patients. In particular, the fact that it is a long-acting injectable administered once every six months can maximize patient adherence.”Sunlenca has already been approved by the U.S. Food and Drug Administration (FDA) and the European Medicines Agency (EMA) and is currently in use. Recently, it has also demonstrated exceptional efficacy as a pre-exposure prophylaxis (PrEP), drawing attention as a potential game-changer, set to reshape the HIV treatment market.The FDA approved Sunlenca in December 2022, following fast-track, breakthrough therapy designation (BTD), and priority review (PR) designations. The EMA approved it earlier in August 2022, and Japan’s PMDA approved it through priority review in August 2023.In Korea, it was designated an orphan drug in December 2024, and on March 4 last year, it was designated as GIFT product No. 46, reflecting the lack of existing treatment options.The indication submitted for its use in Korea is as combination therapy with other antiretrovirals for the treatment of HIV-1 infection in hin adults with multidrug-resistant HIV-1 infection who have a history of treatment failure with current antiretroviral therapy due to resistance, intolerance, or safety issues.
- Policy
- "MOHW will design the drug pricing policy considering NHI cost"
- by Lee, Jeong-Hwan Mar 19, 2026 12:38pm
- Lee Hyung-hoon, Second Vice Minister of Health and Welfare"We recognize that the pharmaceutical industry has varying opinions regarding government policy. However, the government designs policy by looking at where the public's expectations lie and what their interests are. The necessity of fostering the pharmaceutical industry and the fact that domestic generic drug prices are high are areas where society relatively shares a consensus, and social agreement is required. We are not setting a single specific drug pricing reform plan and trying to force it through. We will repeatedly collect and discuss opinions from the pharmaceutical industry. Since policy can change at any time, there is room to modify the reform plan."Lee Hyung-hoon, Second Vice Minister of Health and Welfare (60, Yonsei University), has drawn significant attention by highlighting the difficulties of reconciling diverse interests in a single policy for drug pricing reform, which primarily focuses on lowering the prices of previously-listed generic drugs.Lee stated that, given South Korea's adoption and operation of a single-payer National Health Insurance system, writing a reform plan that simultaneously achieves the goals of fostering the pharmaceutical industry and ensuring efficient, rational national pharmaceutical expenditures requires considering the interests of both the industry and the public as beneficiaries of health insurance.Meeting with the Korea Special Press Association on the 18th, Vice Minister Lee promised, "Rather than taking the stance of a policy authority, the Ministry of Health and Welfare designs policy by examining public interests. However, we have not finished setting a single drug pricing reform plan. Policy is open and can continue to change. We will communicate with the pharmaceutical industry."This was the Vice Minister's response when asked how the Ministry intends to resolve the industry's backlash against the drug pricing system reform plan announced on November 28 last year.Lee stated that, while agreeing on the necessity of fostering the South Korean pharmaceutical and biotech industry on a global scale to strengthen national competitiveness, the final reform plan would be finalized after collecting opposing opinions from the industry on uniform price cuts.The Ministry intends to listen to the industry's voice and to prepare a revised proposal until the Health Insurance Policy Deliberation Committee deliberates on the reform plan on the 26th.Since releasing the initial draft of the reform plan on November 28 last year, the Ministry has presented a revised draft to the industry on March 11 at the subcommittee. However, the domestic pharmaceutical industry continues to criticize the process as 'closed administration,' citing the failure to specify clear generic price calculation rates, mentioning only a range of 'low-to-mid 40%,' the lack of specific timing and methods for price cuts per item, and the absence of detailed criteria and periods for preferential drug pricing.Lee repeatedly stated that he would gather industry opinions before the committee's final decision. He emphasized that, because pharmaceutical expenditures are funded through the health insurance budget, the government views the drug pricing system considering the efficiency of public insurance premiums."South Korea operates the drug pricing system based on the health insurance finances funded by the mandatory premiums paid by citizens," Lee stated. "Because medical fees and drug prices for both generics and new drugs are operated through health insurance finances, we value the National Health Insurance cost, which represents public expectations."Lee also noted that he recognizes the domestic pharmaceutical industry as a partner in South Korea's efforts to advance to one of the top five global pharmaceutical and biopharmaceutical powers. This is interpreted as a necessity for the administration to communicate and make adjustments to the aspects the industry hopes will be reflected in the reform plan.Lee stated, "Since the Ministry of Health and Welfare also aims to leap into a top-five pharmaceutical and bio-powerhouse as a national task, the pharmaceutical industry is an important partner," Lee explained. "We are discussing the reform plan with the industry. There is a backlash because the industry has more favorable, explicit expectations, but we will continue to communicate. There is room for modification within the system."Lee added, "There may be differing opinions because the industry has its own perspective and expected levels. While the Ministry has its position as a policy authority, we work by looking at where the public views, perspective, and interests lie."Lee stated, "We are open to differing opinions on Ministry policy or points raised by the media. We are not setting one thing and trying to force it through," adding, "Policy can continue to change."Lee concluded by saying, "The need to foster the pharmaceutical industry and the fact that generic prices are high are areas where there is a relatively social consensus. We have carefully considered the drug pricing policy. We are listening to the pharmaceutical industry's opinions," adding, "We fully recognize, acknowledge, and respect the necessity for innovative pharmaceutical companies and the domestic industry to possess national competitiveness and move toward becoming a bio-power. We support and share with the industry's aspiration to expand to a larger global stage."
- Policy
- MOHW drug pricing reform to likely bypasses NA briefing
- by Lee, Jeong-Hwan Mar 18, 2026 09:11am
- Joomin Park, chair of NA Health and Welfare CommitteeThere is a growing likelihood that the Ministry of Health and Welfare’s drug pricing system reform plan, the first in 14 years, will be finalized and implemented without a formal briefing to the National Assembly.Although Joomin Park, Chair of the National Assembly Health and Welfare Committee from the Democratic Party of Korea, instructed Health and Welfare Minister Jung Eun-kyung to provide a separate briefing on the reform plan, it has been confirmed that the ruling and opposition floor leaders on the committee are not actively coordinating a schedule for a plenary session.On the 17th, an official from an opposition lawmaker’s office on the National Assembly’s Health and Welfare Committee explained, “We have heard absolutely nothing regarding a plenary session for a separate briefing on the drug pricing reform plan.”If this explanation is correct, it means that despite Chair Park’s remarks on the need for a separate briefing on the drug pricing system, negotiations between Democratic Party floor leader Sujin Lee and People Power Party floor leader Miae Kim have come to a complete standstill. If an additional plenary session for the briefing is not held, the Ministry of Health and Welfare will proceed with the drug pricing reform plan, which has faced backlash from the pharmaceutical industry, without reporting to the National Assembly.The need for an additional National Assembly briefing on the drug pricing reform plan first arose during the plenary session held on the 10th for the annual policy briefing by relevant government ministries, when Rep. Sun-min Kim of the Rebuilding Korea Party raised the need through a procedural intervention.Rep. Kim criticized the ministry for attempting to push forward rapidly with the drug pricing reform—centered on across-the-board generic price cuts—despite opposition from the pharmaceutical industry, while including no update at all on the reform plan among the key issues in its official policy briefing.At the time, Committee Chair Park agreed with Kim and told Minister Eun-Kyoung Jeong, “Because this is a very important matter, it would be a good idea to provide an additional briefing at a plenary session once the procedures related to the reform plan are completed.”Nevertheless, the reason why the convening of the Welfare Committee’s plenary session remains a distant prospect appears to be rooted in the Ministry of Health and Welfare’s passive attitude and the suspension of consultations between the ruling and opposition parties.The Ministry of Health and Welfare has no incentive to take the lead in convening a plenary session at the administrative level, free from the burden of a one-point briefing, as the reform plan to lower the generic drug pricing rate from 53.55% to the 40% range has met with opposition from the pharmaceutical industry.As for the ruling party, given the ongoing fierce criticism from the opposition regarding the national vaccination campaign for COVID-19 vaccines found to contain foreign substances, which proceeded without proper procedure, the party has no reason to hold an additional plenary session that would expose it to further attacks from the opposition.In fact, People Power Party lawmakers on the Legislation and Judiciary Committee have decided to file a complaint accusing Minister Eun-kyoung Jeong, who served as Commissioner of the Korea Disease Control and Prevention Agency during the COVID-19 pandemic, of alleged dereliction of duty over the administration of 14.2 million doses of contaminated vaccines.Time constraints also make an additional plenary session difficult. The ministry plans to finalize the details and implementation schedule of the drug pricing reform at the HIPDC meeting on the 26th, and physically, it may be impossible to hold an additional National Assembly briefing before then.If the additional plenary session of the Health and Welfare Committee fails to materialize, the ministry is expected to have the reform plan approved at the HIPDC without first reporting it to the National Assembly.The Ministry of Health and Welfare plans to finalize the specific details and implementation date of the reform plan at the full HIPDC meeting on the 26th, following a subcommittee meeting on the 11th focused solely on the drug pricing reform plan and another subcommittee meeting on the 18th.An official from an opposition party lawmaker’s office on the Health and Welfare Committee said, “It is uncertain whether a plenary session for an additional briefing on the drug pricing reform plan will be held. Time is tight physically, and I understand that both the ruling party and the ministry are passive about holding one. The conflict between the ruling and opposition parties over the contaminated vaccine issue and calls for Minister Jeong’s accountability are also affecting the failure to agree on the briefing for the drug pricing reform plan.”The official continued, “In a situation where the opposition is demanding a hearing on contaminated vaccines and the indictment of Minister Jeong, an additional briefing on the drug pricing reform plan, which is already drawing strong resistance from the pharmaceutical industry, would naturally be burdensome for the ruling party. Although the pricing reform is an important policy that will shape the future of the pharmaceutical industry, and although Chair Park has requested a report on it, the government and ruling party seem to lack the will to proceed.”An official from the office of Democratic Party floor leader Sujin Lee stated, “Given the timing, it seems difficult to hold an additional plenary session on the drug pricing system, but we plan to hear the opinions of the opposition party’s floor leadership. If an additional briefing proves difficult, we will also consider a plan for ruling party lawmakers to receive individual briefings on the reform plan from the Ministry of Health and Welfare.”
- Policy
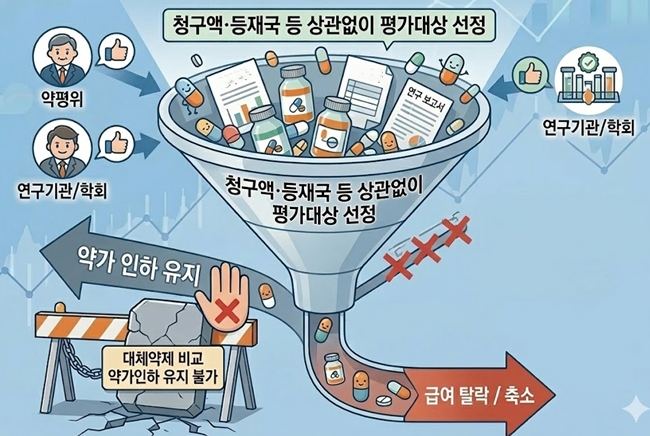
- Scope expanded, outcomes tightened for reimbursement reassessment in Korea
- by Jung, Heung-Jun Mar 18, 2026 09:11am
- The government is moving to specify implementation criteria as part of its efforts to reform the reimbursement adequacy reassessment system. It appears that ingredients for which the Drug Reimbursement Evaluation Committee (DREC), research institutes, academic societies, and others raise the need for reassessment will undergo reevaluation regardless of claim volume or listing-country criteria.In addition, the option of maintaining reimbursement through price cuts by comparing a product with its alternatives is expected to disappear going forward.The government is specifying the implementation criteria for reimbursement adequacy reassessment. AI-generated image.According to industry sources on the 17th, the government is specifying the implementation requirements for reimbursement reevaluation reassessments ahead of deliberation by the Health Insurance Policy Deliberation Committee (HIPDC) later this month.Until now, reassessments have been conducted on ingredients among older listed drugs that met the claim volume and countries of listing criteria.The reform plan announced last November signaled a shift away from criteria based on claim volume and listed countries, instead focusing on: ▲ ingredients for which national health authorities in A8 countries have initiated clinical or reimbursement adequacy reassessment; ▲ cases where data or clinical evidence contradicting previously reported efficacy has been published; and ▲ drugs for which the need for reassessment has been recommended by academic societies or experts.Recently, ingredients deemed necessary by DREC through monitoring, as well as those evaluated by research institutions or public agencies, have been subject to additional review.This means the committee will be able to proactively examine and designate a broader range of candidates for reassessment. As a result, concerns are emerging within the industry that the number of drugs subject to reassessment may increase.A pharmaceutical industry official said, “There had already been a tendency to broaden the criteria and expand the scope of targets. The intent is to cut drug spending by strengthening reassessment. Since the idea is that reassessment will proceed whenever there is a perceived need, I believe the number of targets will likely increase.”In particular, because the results of reassessment are being simplified into either delisting or positive listing, the weight of target selection becomes even greater. The option of comparing a product with alternative therapies and maintaining reimbursement through price cuts will disappear.Another industry official said, “It is difficult to predict how things will unfold next year. Depending on the new implementation criteria, the number of targets could either increase or decrease. However, considering the practical work involved in reassessment, the authorities cannot simply increase the number of ingredients indiscriminately, so the annual number of reassessment targets may not change significantly.”
- Policy
- Prices of top-priced generics could drop 32% upon pricing reform
- by Chon, Seung-Hyun Mar 18, 2026 09:11am
- The government’s announced tiered pricing system for generics is expected to function as a powerful mechanism that significantly lowers generic drug prices. Concerns have been raised that the faster exposure to the tiered pricing system compared to the current system could dampen the momentum for later-entering generics. It is estimated that the price of the 13th generic to enter the market will drop to half the current level, as the discount rate increases for products that fail to meet the highest-price criteria, such as those failing bioequivalence tests.Analysis suggests that even if a product meets the highest-price criteria, if more than 13 products are launched simultaneously, a new price reduction mechanism, which applies an additional 15% price cut one year later, will be triggered, causing the price of the first generic to fall by 32% compared to current levels.Tiered pricing system with 15% reduction applied from the 13th generic onward... Drug prices plummet due to stricter eligibility requirementsAccording to industry sources on the 16th, the Ministry of Health and Welfare presented a principle at the HIPDC subcommittee meeting held on the 11th that, under the price reform, the 13th generic would be subject to pricing. The price reduction rate applied to each tier would be 15%.The price drop for generics become greater with the reformThe tiered pricing system is structured such that the insurance ceiling price decreases on a monthly basis, the later a generic enters the market. Although it was abolished in 2012, the system was reinstated with the 2020 drug pricing reform. Under the current system, if there are more than 20 pre-listed products of the same formulation, the price of generics entering the market as latecomers is reduced by 15% at each step.When reporting the drug pricing system reform to the Health Insurance Policy Deliberation Committee on November 11 of last year, the Ministry of Health and Welfare proposed a policy to grant the first generic a price reduced by 5 percentage points (p) from the calculated price starting from the 11th listing of the same formulation. This effectively means the Ministry presented a relaxed version of the stepped pricing system just four months after its initial report.However, because the tiered system would now begin applying from the 13th generic instead of the 21st under the current system, products would be exposed much earlier to this additional price-cutting mechanism.The impact of the tiered pricing system becomes even greater as the generic pricing benchmark itself is lowered. The government has proposed lowering the benchmark for calculating generic drug prices from the current 53.55% to the low to mid-40% range, a reduction of about 10 percentage points. If the generic drug pricing benchmark drops from 53.55% to 43%, the maximum generic price would be reduced by 19.7% in absolute terms.For example, under the current drug pricing system, when the maximum generic price is KRW 53.55, the 21st generic’s price cannot exceed KRW 45.52, which is a 15% reduction. The price of 22nd and 23rd generics then drops to KRW 38.69 and KRW 32.89, respectively. The 24th generic would be KRW 27.95, and the 25th would be KRW 23.76, meaning drug prices decrease as generics enter the market later.Under the revised pricing system, where the generic drug pricing benchmark is set at 43%, if the maximum price is KRW 43, the price of the 13th and 14th generics drops to KRW 36.55 and KRW 31.07, respectively, following a 15% step-down reduction. The price of the 15th generic drops to KRW 26.4. Under the current drug pricing system, the 15th generic drug is not subject to the tiered pricing structure and can maintain a price of 53.55%, but under the revised system, its price drops to less than half that level.As the stricter maximum price requirements under the revised system will also be applied, the magnitude of price reductions under the tiered pricing system will become even greater.Since July 2020, under the revised generic pricing rules, a generic product must both conduct its own bioequivalence study and use a registered active pharmaceutical ingredient to qualify for the top price. For each unmet requirement, the ceiling price is reduced by 15%. If both requirements are not met, the price falls by 27.75%. Applying that 15% reduction, the 53.55% premium benchmark falls to 45.52% if one requirement is unmet and to 38.69% if both are unmet.According to the criteria introduced by the ministry after the 2020 pricing reform, “Even if a product meets both premium-price requirements, if there are already 20 or more listed identical products, starting from the 21st product, the price will be listed at 85% of the lower of the lowest price of the identical product or 38.69%.” The 38.69% rate is the result of two 15% reductions applied when a product failed to meet both of the highest-price criteria. (53.55% × 0.85 × 0.85)When 20 or more generics are listed, the 21st drug is listed at the lowest price vs 38.69%, whichever is lowerCurrently, the 21st generic drug,the first to be subject to the tiered drug pricing system, is priced at 32.86%, which is a 15% reduction from the 38.69% baseline. Compared to the highest price of 53.33%, this means the first generic drug subject to the tiered pricing system will see a 38.6% reduction. The prices of the 22nd and 23rd generic drugs will be reduced even further.The Ministry of Health and Welfare plans to increase the reduction rate applied when the highest price requirement is not met under the revised drug pricing system from 15% to 20%. If the benchmark for the highest generic drug price is set at 43%, the calculation standard will be further lowered to 34.40% for generics that fail to meet one requirement, and to 27.52% for generics that fail to meet both requirements.Under the revised drug pricing system’s tiered pricing structure, which applies the highest-price requirement, the ceiling price drops significantly starting with the 13th generic.The price of the 13th generic is calculated to drop from 27.52%, the rate applied to generics failing to meet two highest-price requirements, to 23.39%, a reduction of 15%. Comparing the same 13th generic drug, the price under the current system is KRW 53.55, whereas under the revised system, it drops to roughly half that amount. The 13th and 14th generic drugs each decrease by 38.6% from the lowest price, falling to KRW 14.36 and KRW 8.8, respectively.Under the revised drug pricing reform, the combined effects of shortening the sequence for applying the tiered pricing system, a 15% reduction upon tiered application, and a 20% price reduction for drugs that do not meet the highest-price requirement create a structure that effectively prevents late-entrant generics from entering the market.If 13 or more highest-priced generics are listed, prices will be reduced by 15% after one year.Even top-priced generics could see their prices cut a year later, depending on the number of products listed at the same time.To prevent excessive competition when the first generics enter, the ministry plans to apply pricing standards equivalent to tiered cuts to generics whose listing causes the total number of identical products to exceed 13.Even if a generic is listed within the first 12 products and initially receives the top price of 43%, if that product is among a group whose simultaneous entry causes the total to exceed 13, its price would be cut by 15% one year later.The price of generics with many listings will fall to a greater extent with the new reformFor example, if 8 generic products are listed in January and receive prices in the low-to-mid 40% range, and another 8 generics are listed in February, those products would all be treated as being listed simultaneously starting from the 9th position and could receive the top price. However, because the additional eight February listings push the total number of identical products beyond 13, those products would, one year later, fall to the 85% tiered level.Even the earliest listed generic could have its price cut by 15% one year later if 13 or more products enter simultaneously. In other words, even if it initially secures the top-price benchmark of 43%, it could later fall to 36.55% after one year. In that case, the price would be 31.75% lower than the current ceiling price.Within the industry, this is being interpreted as the government effectively intending to allow only up to 12 generics. Under restrictions on joint development, only three products may participate in one bioequivalence study. Generics beyond three such groups would face such sharp price declines that their incentive to enter the market would effectively collapse.One industry official noted, “Once the revised drug pricing system is implemented, late-entering generics will effectively be unable to turn a profit, so competition to secure the highest price by capturing the market first will inevitably intensify. Even if a company secures a leading position in the generic market, if there are many competing products, the tiered pricing mechanism will cause drug prices to fall, which could even lead to companies abandoning their plans to enter the generic market altogether.”
- Policy
- Boehringer discontinues original meloxicam 'Mobic Capsules' in KOR
- by Lee, Tak-Sun Mar 17, 2026 09:22am
- Product photo of 'Mobic Capsules''Mobic Capsules,' the original drug of the Non-Steroidal Anti-Inflammatory Drug (NSAID) meloxicam, is set to discontinue its supply in South Korea.Boehringer Ingelheim's headquarters has decided to stop imports, with product stocks expected to be depleted by April next year. The decision to withdraw from the Korean market is interpreted as a result of low sales performance and a market saturated with numerous alternatives of the same ingredient and class.According to industry sources on the 16th, Boehringer Ingelheim Korea has notified distributors regarding the discontinuation of Mobic Capsules.Boehringer Ingelheim stated, "The company has decided to discontinue the supply after recognizing that a wide variety of alternative treatments are already available in the Korean market." The company added, "Currently imported stocks will be supplied until they are exhausted, noting that the expected depletion date may vary depending on usage volume. The projected depletion for Mobic Capsules 7.5mg is around April next year, while Mobic Capsules 15mg is expected to run out by March next year."Mobic is an NSAID containing meloxicam as its active ingredient. It is effective in alleviating inflammation and pain associated with rheumatoid arthritis, osteoarthritis, and ankylosing spondylitis by selectively inhibiting COX-2. While it tends to have fewer gastrointestinal side effects compared to traditional anti-inflammatory drugs, it carries risks of blood clots and cardiovascular issues; therefore, it should be taken at the lowest effective dose for the shortest duration possible.Currently, there are 82 meloxicam products in South Korea at the 7.5mg dose and 15 at the 15mg dose. Even with the withdrawal of the original brand, the impact of the supply disruption is expected to be minimal due to the abundance of substitute medications.Mobic's sales have also been decreasing. According to UBIST, its outpatient prescription sales last year were KRW 1.5 billion, a 2% decrease from the previous year. This is a significant gap compared to Celebrex, another drug in the same NSAID class, which recorded KRW 41.7 billion in outpatient prescriptions last year.Consequently, Boehringer Ingelheim may have decided to halt supply to focus on other pharmaceutical products, given Mobic's low profitability in the Korean market.
- Policy
- Generics companies challenge ₩100B Lixiana·Livalozet mkt
- by Lee, Tak-Sun Mar 17, 2026 09:22am
- LixianaGeneric drug development is accelerating for Lixiana (edoxaban tosylate hydrate, Daiichi Sankyo Korea) and Livalozet (pitavastatin calcium + ezetimibe, JW Pharmaceutical), both of which are approaching the end of their exclusivity periods.Because both drugs have recorded more than KRW 100 billion in prescription sales, they are emerging as major targets for generic manufacturers this year and next.In the case of Lixiana, it has been disclosed that 13 products have applied for approval this year alone.According to the Ministry of Food and Drug Safety on the 16th, under the approval-patent linkage system, the original company was notified of marketing authorization applications filed for 13 generic products containing edoxaban tosylate hydrate in 2026.The recent increase in Lixiana generic approval applications is due to the fact that its substance patent is scheduled to expire on November 10 of this year.As for the pharmaceutical composition patent, which is scheduled to expire on August 21, 2028, most generic manufacturers have already succeeded in circumventing it through passive scope-of-rights confirmation trials. Once the patent expires, there will be no obstacles to launching generic drugs on the market.Lixiana is a direct oral anticoagulant (DOAC) whose prescription volume has continued to expand as it replaces warfarin. According to UBIST, its outpatient prescription sales alone reached KRW 117.5 billion last year.Generic companies had made attempts to invalidate the substance patent due to its strong commercial value, but failed. In addition to the original product, nine follow-on companies have already obtained product approvals, and their generic versions are expected to be launched after the substance patent expires.Since no follow-on product has secured first generic exclusivity, it appears that many products will flood the market simultaneously upon expiry of the substance patent, without restrictions on sales.LivalozetThere is also a strong possibility that a large number of generics will emerge for Livalozet, which posted KRW 117 billion in outpatient prescription sales last year. On the 13th, Nelson Korea, Pharmbio Korea, and Austin Pharmaceuticals all received approval on the same day for their bioequivalence study protocols for generic development.Generic development has already begun in earnest since 2024, with many pharmaceutical companies aiming to file applications. Applications can be submitted after the Livalozet re-examination period ends on July 27 next year.Livalozet is a dyslipidemia treatment combining pitavastatin and ezetimibe, and it has gained popularity due to its lower risk of diabetic side effects and strong lipid-lowering efficacy.In addition to the original company, JW Pharmaceutical, 5 companies have directly launched products with the same active ingredients through their own clinical trials. Ahn Gook Pharmaceutical, Boryung Pharmaceutical, Dongkwang Pharmaceutical, Hanlim Pharm, and Daewon Pharmaceutical launched related products in the second half of 2023 and have continued rapid growth. Last year, Ahn Gook Pharmaceutical’s Pevarozet recorded KRW 29.2 billion in outpatient prescription sales, while Daewon’s Tavalozet recorded KRW 18.2 billion. These companies also succeeded in launching their products early by invalidating Livalozet’s use patent.More recently, low-dose products combining pitavastatin 1 mg and ezetimibe 10 mg have also appeared. In January, Ilsung IS, Ildong, Daewoong, and Hanlim obtained approvals for related products, and this month, JW Pharmaceutical also received approval for a new product.Given recent sales trends, there is a high likelihood that generic drugs will flood the market once the re-evaluation period ends next July.
- Policy
- The key elements of government’s drug pricing reform plan
- by Jung, Heung-Jun Mar 17, 2026 09:22am
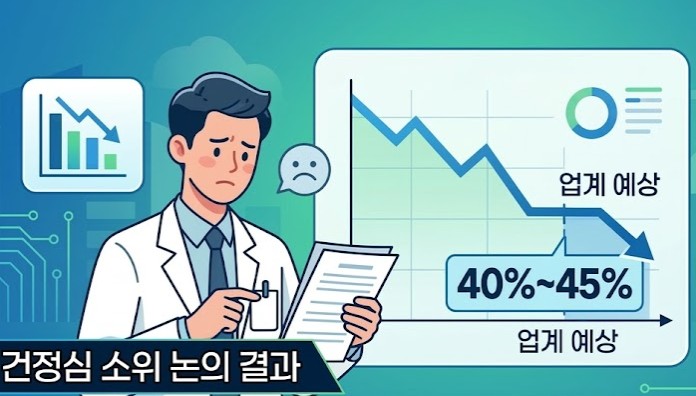
- Under the government’s new drug pricing reform, price cuts for already listed drugs are expected to begin in the third quarter, with preferential treatment for innovative companies’ new listings expected to begin next year.Innovative companies are set to receive a temporary exception from price cuts for existing drugs. However, the duration of these exceptions is scheduled to be finalized at the Health Insurance Policy Deliberation Committee meeting on the 26th.In addition, the fast-track listing–post-evaluation and adjustment track is planned for 2028, and its criteria will be established to select which products are eligible.According to industry sources on the 16th, the Ministry of Health and Welfare plans to finalize the drug pricing reform at the HIPDC plenary meeting on the 26th. Based on the discussions at the subcommittee, the direction of the reform will be fixed after some further adjustments.AI-generated image◆ Generic pricing ratio in low-to-mid 40% range…failure to meet required criteria: 85% → 80% = The pricing ratio, which had been described as “in the 40% range” last November, has recently narrowed to the low-to-mid 40% range at the HIPDC subcommittee.The industry expects it to fall somewhere between 43% and 45%. Since many had hoped for the high 40% range, the industry had expressed disappointment. However, because time remains before the HIPDC meeting, the final rate still remains to be seen.If the required conditions for generic pricing are not met, the pricing level is expected to be lowered from 85% to 80%. If a company does not submit its own bioequivalence data or fails to use APIs registered with the MFDS, the reduction rate will become greater.◆ Price cuts for already listed drugs to begin in Q3…cuts will be deferred for innovative companies, but duration undecided=The government plans to begin price cuts for already listed products in Q3. It intends to divide companies into groups based on their listing dates and implement sequential adjustments using varying calculation rates.Innovative pharmaceutical companies will be granted a deferral period under a separate pricing calculation rule. However, an exception is being discussed for ingredients with 21 or more listed products. A deferral period of 4 to 5 years has been mentioned, but it will be finalized at the HIPDC meeting on the 26th.For innovative companies, their reduction ratio under the price-volume agreement will be raised from 30% to 50%, with application expected to start at the end of this year.◆ New “innovation-equivalent” category to be established next year…50% preferential pricing=When newly listing drugs, innovative companies will receive a 60% pricing add-on. The period for this premium will be 1+3 years. If the product is manufactured domestically, an additional 3 years will be granted.In addition, the government is considering adding a category of companies equivalent to innovative firms, with 50% preferential pricing. These “quasi-innovative” companies would be defined based on annual sales above or below KRW 100 billion, with R&D investment ratios of at least 5% or 7% of sales, respectively.However, companies that have received administrative sanctions for rebates within the past 5 years will be excluded. The preferential period for quasi-innovative companies is expected to be the same as that for certified innovative companies.The preferential measures for innovative and quasi-innovative companies are expected to take effect starting next year.◆ Stepwise price reduction from the 13th product onward…15% reduction rate maintained=To manage multiple generic listings, the government plans to strengthen the stepwise reduction system. The stepwise reduction, which currently begins from the 21st product, is expected to shift to the 13th listed product.The reduction rate itself is expected to remain unchanged, with the price set at 85% of the previous lowest price. However, if multiple products are listed at the point when the 13th product is entered, the price of those products will be adjusted to the 85% level one year later.The strengthened stepwise reduction and multi-product listing management are expected to take effect next year.◆ 100-day fast-track listing to be institutionalized next year…selection criteria for ‘innovative new drugs’ to be established=The government plans to institutionalize fast-track listing for innovative new drugs next year. It also plans to establish post-evaluation and price-adjustment mechanisms.Selection criteria will also be established for determining which innovative new drugs will qualify for fast-track listing. The operation of a grading system is also under review.The government plans to operate a performance-based evaluation model incorporating digital healthcare elements such as hospital EMR systems and AI data, and is also reviewing the creation of a specialized agency.It appears that decisions to delist drugs will be made through post-market evaluation. The plan is to establish an evaluation and adjustment system to determine selective reimbursement coverage and appropriate drug prices.◆ Flexible drug pricing contract system to expand in Q2…will prepare measures to prevent inconvenience to patients =The expansion of the so-called ‘flexible drug pricing contract system,’ which allows for contract prices different from the listed price, is expected to be implemented in Q2.The expanded scope is expected to include listed new drugs, already listed off-patent originals, new drugs whose RSA refund period has ended, incrementally modified new drugs, and biosimilars.As the number of products subject to dual pricing increases, the government is preparing measures to prevent inconvenience to patients. One approach under review is to charge patients based on the separately contracted price rather than the listed price, thereby preventing the need for refunds.◆ Price evaluation and adjustment every 3–5 years…targeting ingredients 5 years after first generic entry=The government is establishing a system for evaluating and adjusting drug prices every 3–5 years. This involves comparing, by ingredient, ▲the number of products, ▲market structure, and ▲drug prices in major countries.The target appears to be ingredients for which 5 years have passed since the first generic entered the market. The specific operational model will be established and implemented after gathering industry feedback.The Ministry of Health and Welfare is reportedly expected to revise and supplement the reform plan containing these elements and approve it at the HIPDC meeting on the 26th.
- Policy
- Approval of HER2-bispecific Ab 'zanidatamab' in KOR imminent
- by Lee, Tak-Sun Mar 16, 2026 09:25am
- BeOne Medicines' 'zanidatamab (product name: Ziihera 300mg),' a HER2-targeted bispecific antibody, is likely to be approved soon.Zanidatamab is the first HER2-targeted therapy for biliary tract cancer (BTC) approved by the U.S. FDA. Once it is introduced to the Korean market, this drug is expected to improve treatment outcomes for local patients significantly.According to industry sources on the 12th, the Ministry of Food and Drug Safety (MFDS) recently completed the safety and efficacy evaluation for zanidatamab. Once a "suitable" decision is made from this review, only the final administrative granting of the license remains. Furthermore, under the "Approval-Reimbursement Linkage System," the company can apply for insurance coverage through the Health Insurance Review and Assessment Service (HIRA) immediately following the completion of the safety/efficacy review.In November 2024, the U.S. FDA granted accelerated approval of zanidatamab for patients with previously treated HER2-positive advanced or metastatic BTC. There has been a high unmet medical need in HER2-positive BTC patients.Treatment options for HER2-positive BTC have been extremely limited. BTC is known for its poor prognosis, with a 5-year survival rate of less than 5% once metastasized. In the pivotal HERIZON-BTC-01 clinical trial, the zanidatamab group showed an objective response rate (ORR) of approximately 52%, and a median duration of response (DOR) of approximately 14.9 months.Following its FDA approval, launch in Korea was expedited after the drug was selected for the Global Innovative products Fast Track (GIFT) program in December 2024. In Korea, the specific indication was submitted for approval for the treatment of adult patients with previously treated, unresectable, locally advanced, or metastatic HER2-positive (IHC 3+) biliary tract cancer.Unlike conventional monoclonal antibodies, zanidatamab is a bispecific antibody that binds simultaneously to two distinct sites on the HER2 (Human Epidermal Growth Factor Receptor 2) protein. This dual-binding mechanism allows for more potent inhibition of cancer cell growth.Beyond BTC, clinical development is currently expanding into gastric and gastroesophageal junction (GEJ) adenocarcinomas.BeOne Medicines underwent a global corporate rebranding last year. Beigene Korea was originally a China-based oncology developer; the company later changed its name to BeOne Medicines and moved its legal registration to Switzerland. In Korea, the company already supplies innovative oncology drugs, including Brukinsa (zanubrutinib) and Tevimbra (tislelizumab).Ziihera is a new drug originally developed by Jazz Pharmaceuticals. BeOne Medicines holds the commercialization rights for the Asia-Pacific region.
- Policy
- Bill proposed to apply 5% coinsurance for follow-up tests
- by Lee, Jeong-Hwan Mar 16, 2026 09:25am
- An amendment to the National Health Insurance Act is being proposed to reduce the patient coinsurance rate to 5% for follow-up examinations for patients with cancer, rare diseases, and severe or intractable illnesses.A bill to amend the Patient Safety Act has also been introduced in the National Assembly. It imposes an obligation on doctors to fully explain the details and circumstances of medical accidents to patients, while stipulating that expressions of consolation, empathy, or regret made by doctors during this process cannot be used as evidence of the doctor’s liability or to their disadvantage in medical malpractice lawsuits.On the 13th, Rep. Kyo-heung Kim of the Democratic Party and Representative Cheol-soo Ahn of the People Power Party each introduced a bill to amend the National Health Insurance Act and the Patient Safety Act, respectively, containing these provisions.Bill to reduce patient coinsurance for follow-up tests for serious diseases to 5%Representative Kim’s bill proposes reducing the patient coinsurance rate to 5% for follow-up examinations for cancer, rare diseases, and severe or intractable diseases that have a high risk of recurrence.Under the current law, insured individuals and their dependents are required to bear part of the cost of medical services when receiving healthcare benefits.For certain diseases with high treatment costs, a special calculation system is applied in which the patient’s share of medical costs is reduced to 5–10% of the total medical benefit costs for a specified period.Rep. Kim pointed out that while cancer, rare diseases, and severe or intractable diseases carry a high risk of recurrence even after treatment, making continuous monitoring and follow-up examinations essential, patients face high testing costs once the special calculation exception ends after the disease is cured.He argued that this creates a problem where patients are unable to receive proper follow-up examinations due to such a financial burden.To address this, accordingly, Representative Kim introduced a bill that reduces the patient cost-sharing rate to 5% when cancer patients, rare disease patients, or patients with severe or intractable diseases undergo follow-up examinations for their respective conditions.Bill preventing doctors’ expressions of sympathy after medical accidents from being used as evidence in lawsuitsRepresentative Cheol-soo Ahn introduced a bill requiring heads of healthcare institutions or healthcare professionals to make efforts to sufficiently explain the details and circumstances of patient safety incidents. The bill also stipulates that expressions of consolation, empathy, or regret made by doctors during this process cannot be used as evidence regarding the doctor’s liability in medical malpractice lawsuits or other legal proceedings.Under the current law, there is no separate provision requiring doctors to explain the details or circumstances of patient safety incidents.In particular, doctors sometimes avoid providing explanations or maintain a defensive stance regarding patient safety incidents, as expressions of humanitarian regret or explanations could later be used as evidence of the healthcare institution’s or healthcare professional’s liability in future trials related to the incident.Representative Ahn expressed concern that the information gap and lack of communication between doctors and patients deepen distrust among patients and their guardians toward healthcare institutions, turning simple incidents into complex legal disputes.He also noted that this situation creates excessive psychological pressure and litigation burdens on healthcare professionals, contributing to instability in the medical environment.The proposed bill would therefore require doctors to provide detailed explanations about medical accidents, while explicitly stating that expressions of sympathy, empathy, or regret cannot be used as evidence of liability in medical lawsuits.Rep. Ahn explained, “This bill aims to restore trust between healthcare professionals and patients and minimize unnecessary social costs, such as those arising from litigation.”