- LOGIN
- MemberShip
- 2026-07-23 05:46:00

- Policy
- The fast track of tx for serious dz without a substitute
- by Lee, Tak-Sun Jan 03, 2023 05:40am
- Prior consultations will be newly established before the main negotiation to quickly register anticancer drugs without alternative drugs and treatments for severe and rare diseases. Accordingly, the negotiation period for the drug will be reduced from 60 days to 30 days. The NHIS announced on the 30th some revisions to the drug price negotiation guidelines containing such information. According to the amendment, among PE drugs, drugs that are evaluated as RSA (Expenditure Cap drugs or Refund) will have a negotiation period of 30 days. These drugs, etc. can be consulted in advance before the main negotiation if ordered by the Minister of Health and Welfare. Accordingly, the target drugs may undergo prior consultation by providing data to the NHIS 15 days before submission to the HIRA Drug Benefit Evaluation Committee to determine their benefit adequacy. Therefore, in this negotiation, the negotiation period will be reduced from 60 days to 30 days. The NHIS has decided to apply the guidelines from January 1 next year. Earlier, the HIRA also supported the introduction of a preliminary consultation system by specifying drugs that can omit submission of economic evaluation data through the "revision of regulations on evaluation criteria and procedures such as whether drugs are eligible for medical care benefits." According to the revision, the standard for PE omission drugs is established that "the target patients are few." There are no products or treatments with equal therapeutic positions as drugs used in children, and cases of clinically significant improvement in quality of life have been added to be recognized by the committee. As Canada is included in the drug price reference country, PE can be omitted for drugs that are publicly paid in more than three of the eight foreign countries (Japan, France, Germany, Italy, Switzerland, the United Kingdom, the United States, and Canada). This was also applied to the detailed evaluation criteria for drugs subject to negotiation, such as new drugs, which are the HIRA internal guidelines. All of them will go into effect on January 1 next year.
- Policy
- Will Koselugo, a new neurofibroma drug be reimbursed next ye
- by Lee, Tak-Sun Jan 03, 2023 05:40am
- "Koselugo (Selumetinib, AstraZeneca), the first drug used for childhood neurofibromatosis, a rare disease, is stepping up its challenge." Although he had a hard time at the HIRA in March, efforts to register health insurance have continued since then, such as supplementing data and reapplying for benefits. Starting next year, expectations for the registration are growing as the drug price negotiation period for drugs designated as drugs subject to rapid screening, such as Koselugo, is shortened. According to the industry on the 30th, Koselugo has continued its efforts to register benefits by submitting supplementary data since it was non-reimbursement in March. Since then, the HIRA has also conducted a review by listening to related academic opinions and reviewing standards. It is known that Koselugo's existing drug decision application was recently withdrawn and a new drug decision application was submitted. Koselugo is interpreted as a willingness to continue its efforts to register benefits next year. Starting next year, drugs designated by the Ministry of Food and Drug Safety as life-threatening or critical treatments such as Koselugo will speed up benefit screening and negotiations. The HIRA and the NHIS recently revised related guidelines to provide data to the NHIS in advance for drugs used for life-threatening diseases from next year, moving the negotiations forward by about 30 days. As a result, if Koselugo retries and passes the drug evaluation committee, the pace of registration is expected to accelerate. Patients' opinions on Koselugo are also greater than ever. Neurofibroma has relied on symptomatic treatment without proper treatment. About half of Type 1 patients experience PN, which can occur anywhere in the body along the nerves, and the range of motion is limited depending on the location and size of the nerve or causes pain and appearance problems. If a tumor develops inside, it compresses the internal organs, and most of the tumors are positive and grow slowly, but some are malignant or are likely to lead to breast cancer in women. The prevalence is around one in 3,000 people. Koselugo achieved the primary evaluation index ORR by reducing tumor size by more than 20% in 68% of administered patients in clinical trials. In addition, 82% of patients who showed partial reactions continued to respond for more than 12 months. Half of the patients who did not receive treatment suffer from disease progression after 1.5 years, and only 15% of the patients who used Koselugo developed the disease up to 3 years ago. As the drug approaches 200 million won a year, it is urgent to register health insurance benefits to reduce the economic burden on patients. Insurance authorities are cautious about analyzing cost effects as high drug prices have a significant impact on their finances.
- Policy
- Largest-ever drug pricing reeval to be conducted this year
- by Kim, Jung-Ju Jan 02, 2023 06:04am
- The health and pharmaceutical industry is busy preparing various systems and policies for the new year. The reimbursement adequacy reevaluations for listed drugs that had been initiated as a pilot project on choline alfoscerate products settled as an annual policy project and are expected to start full-scale reevaluations on previously notified products. High-priced drugs for pediatric patients will now be eligible for pharmacogenomic evaluation data exemptions (PE exemptions) and subjects for the special calculation system will also be expanded. In addition, the 3rd Comprehensive Plan to Foster the Pharma and Bio Industry will be initiated, and the task force for the development of the service industry that includes the healthcare industry will also be launched within the month and impact the medical and pharmaceutical industry. The National Assembly will discuss the abolition of the sunset clause on state funding for national health insurance and legislation for public late-night pharmacies. Dailypharm prepared a summary of notable changes in the system as well as policy projects that will be made in 2023 in the pharmaceutical and biopharmaceutical industry. First, the conversion factor for medical care facilities will be raised with the start of the new year. The increase rate is 1.6% for hospitals, 2.1% for clinics, 3.0% for oriental medicine hospitals, and 3.6% for pharmacies. Along with this, the health insurance rate will also rise to 7.09% based on those employed. For drug pricing coverage and control, high-priced drugs for pediatric patients may be allowed PE exemptions starting this month. PE exemption may be allowed for drugs used to treat pediatric patients that are therapeutically equivalent or has no available treatment option; and demonstrates improvement in quality of life or are otherwise approved by the committee. The adjustment in the reference countries used to calculate the foreign adjusted drug price that raised concerns in the pharma and bio industry has been made. The government added Canada to the existing A7 (Japan, France, Germany, Italy, Switzerland, the UK, and the US) to A8. Australia, the country that the industry worried would be included as a reference country, was excluded. The reimbursement reevaluations planned for the year are expected to be the largest ever. The government announced that HIRA will be reevaluating reimbursement on a total of 8 ingredients - Rebamipide, Limaprost alpha-cyclodextrin, oxiracetam, acetyl L carnitine, loxoprofen sodium, levosulpiride, epinastine hydrochloride, sodium hyaluronate eye drops. When adding the efficacy reevaluation set to be conducted on sterepto drugs that received conditional approval for 1 year, the number of drugs subject to reevaluation is increased to 9. The discount rate for the drugs will be finalized in the coming December. In February this year, ingredients subject to reimbursement reevaluations in 2024 will be decided upon and reviewed by the Health Insurance Policy Deliberation Committee. Also, a performance evaluation procedure and management plan for high-priced drugs such as Kymriah and Zolgensma, the so-called 'one-shot treatments', will be prepared within the month. Also, the negotiation period for drugs eligible for the Risk Sharing Agreement (RSA) among PE exemption drugs will be shortened by 30 days through the introduction of a prior discussion system, improving accessibility. Also, the scope of drugs eligible for special calculations will be expanded. A total of 42 new rare diseases and artificial kidney dialysis patients with chronic renal failure will also be applied to special calculations. Also, the administrative dispositions used for the drug serial number reporting system will be raised. From January, HIRA will raise the serial number reporting rate standard for administrative dispositions from 85% to 90% at the time of shipment by wholesalers (including manufacturers and importers that supply third-party licensed items). Therefore HIRA will request administrative dispositions to be made to local governments if the reporting rate becomes less than 90%. Also, the ‘ Regulation on Manufacture and Sales Management of Biological Products, Etc.’ that applied to all biological products will not be categorized into 3 product types. The MFDS plans to make the amendments to the regulation before the 17 of this month. The 3rd Comprehensive Plan to Foster the Pharma and Bio-Industry which had been set as a 5-year project will be implemented this year to support all areas ranging from R&D to business development, investment exports, job creation, and institutional infrastructure. The 'Service Industry Development TF', which sets policy directions to revitalize the service industry, including the field of healthcare, will also be launched in earnest this month. The Ministry of Economy and Finance that oversees the project plans to disclose its 5-year plan for innovating the service industry in March through the TF. During the H1 of the year, a pilot project for the ‘Approval Evaluation-Negotiation Linkage System' will be carried out, and subject drugs will simultaneously undergo three tracks of the drug approval process – MFDS’ safety and efficacy evaluations for marketing authorizations, HIRA’s reimbursement adequacy evaluations, and NHIS’s drug pricing negotiations. In the same period, the National Assembly will discuss whether to abolish the sunset clause for state support of health insurance finances and whether to legislate public late-night pharmacies. Although the contract sales organization (CSO) reporting system is expected to be discussed at the plenary session in the H1 of the year, as the effective date for the law is 1 year and 6 months after the promulgation, its actual application is expected to be possible only in the 2024 2H at the earliest, even if the revisions are made rapidly.
- Policy
- SK Chemicals Riluzole is listed
- by Lee, Tak-Sun Jan 02, 2023 06:04am
- Only tablet type is available for Riluzole, and suspension type is expected to be an alternative for patients with difficulty in swallowing. According to industries on the 1st, SK Chemical's Teglutik Suspension was listed at 134,970 won per bottle. Riluzole, the main ingredient of Teglutik, is used in the treatment of Amyotrophic Lateral Sclerosis (ALS), which is known to delay symptoms by several months. Riluzole is the first drug approved for Lou Gehrig's disease and is most commonly used. There are two products in Korea: Rilutek and Yooritek by Riluzole. Based on IQVIA in 2021, Rilutek showed sales of 3.7 billion won and Yuritech 2.5 billion won. Despite such identical products, Teglutik is introducing them to the domestic market because there is a demand for suspension. Patients with Lou Gehrig's disease have early symptoms of weak tongue and neck muscles, making it difficult to chew and swallow. It was difficult for patients who felt these symptoms to take two tablets of Riluzole daily. Teglutik is a suspension that can be easily taken through oral injections. Patients who are difficult to take directly can be injected through a PEG tube. The drug was developed by the Italian pharmaceutical company Italco Pharma and approved by the U.S. Food and Drug Administration (FDA) in 2018. SK Chemicals has completed patent registration since it was introduced in Korea. The upper limit of the benefit 134,970 won per bottle, is 15 days' worth, which is the same as the existing refining price (4,499 won per day) when converted into daily doses. Since there is no price difference, Teglutik is expected to be useful for patients with symptoms of dysphagia. On the other hand, Sanofi and Yoo Young, which divided the market, are expected to focus on protecting the market this year because they have difficult competitors.
- Policy
- High conc Ultomiris Inj 100mg/mL also approved in Korea
- by Kim, Jung-Ju Jan 02, 2023 06:04am
- Handok received marketing authorization for the orphan drug ‘Ultromiris 100mg/mL(ravulizumab)’ that is used to treat Paroxysmal nocturnal hemoglobinuria (PNH) and atypical hemolytic uraemic syndrome (aHUS) and made a step towards supplying the drug in Korea. On the 28th, the Ministry of Food and Drug Safety announced that it had granted marketing authorization for the drug. The drug is a humanized monoclonal antibody (mAb) that specifically binds to the C5 protein and inhibits complement-mediated inflammation and hemolysis, etc. More specifically, the drug is indicated for the treatment of adults with paroxysmal nocturnal hemoglobinuria (PNH), and as a treatment for adult and pediatric patients with atypical hemolytic uraemic syndrome (aHUS) and inhibits complement-mediated thrombotic microangiopathy (TMA). The MFDS explained that the product approved this time is a high-concentration version of the already-approved Ultomiris Inj and has the benefit of being able to reduce the IV infusion time in patients. The MFDS said, “We will continue to make efforts to allow for the prompt provision of treatments with confirmed safety and efficacy based on regulatory science."

- Policy
- Australia, excluding drug price reference countries
- by Lee, Tak-Sun Jan 01, 2023 10:40pm
- Australia's addition to the drug price reference country, which faced opposition from the pharmaceutical industry, failed. The HIRA initially decided to take a step back from adding Australia and Canada to the drug price reference country and add only Canada to the reference country. The HIRA released the "Detailed Evaluation Standards for Drugs Subject to Negotiation, including New Drugs" on the 28th on the work portal of nursing institutions. The evaluation criteria include the contents of PE drugs announced by the HIRA in August and the expansion of drug price reference countries announced in November. According to the revised detailed evaluation criteria, Canada was included in the existing countries of Japan, France, Germany, Italy, Switzerland, the United Kingdom, and the U.S. A7 countries, making it A8. Australia, which was announced in November, was not included in the reference country. In the case of Australia, the domestic pharmaceutical industry strongly protested, saying that the price of generic drugs could be lowered if they are used for re-evaluation of post-registration materials due to low generic drugs. The KRPIA also issued a statement of opposition, fearing that the price of new drugs would be lowered. The HIRA includes Canada, which is acceptable to all stakeholders, to secure the validity of the amendment, and Australia is eventually excluded. The HIRA official said, "Australia was added to the drug price reference country due to similar geographical access and economic conditions, but as the pharmaceutical industry's opinion, we decided to exclude it in terms of industrial similarity." If it is possible to omit the submission of PE data, it will be evaluated as the lowest among the national adjustment prices of eight foreign countries of similar drugs when registered in more than three A8 countries. In addition, even if it is difficult or possible to select foreign similar drugs, 10% of the highest price of alternative drugs is added to the list of less than A83 countries, and the adjustment price of countries excluding similar drugs is used for evaluation. The drug A8 is also referred to in RSA drugs. The detailed evaluation criteria also reflected that "a small number of target patients" were among the requirements for the omission of submission of PE data. The number of patients is judged based on the expected number of patients subject to benefits (in Korea) of the indication, but the current status of the expected number of patients at the time of evaluation of the drug, which has been considered essential for treatment, is considered. For drugs that can be used for both adults, the submission of PE data can be omitted only if the main indication is children. This reflects the HIRA's prediction in August. In this regard, there were many objections, saying that the PE exemption system has retreated to limit the number of patients and exempt PE only from pediatric indication treatments. However, the HIRA applied the original plan as it is, saying that these regulations can be flexibly applied in the evaluation process.
- Policy
- New formulations of narcotics will be as strictly reviewed
- by Lee, Jeong-Hwan Dec 30, 2022 06:33am
- Regulations on narcotics for medical use, such as narcotic appetite suppressants and propofol, which is used for general anesthesia, that the government is restricting new approvals for, are expected to become stricter than before. Until now, even narcotic medications for which new approvals are restricted by the government were allowed to receive new approvals when developed into new formulations, but these new formulations will not be approved in the future if they have a high risk of misuse or abuse or is rejected by the deliberation committee. On the 19th, the Ministry of Food and Drug Safety announced that it had publicized the narcotic medications subject to restrictions that contain the restrictions above. More specifically, the MFDS had made the notification on the 28th of last month, upon which the notice immediately took effect. The drugs subject to restrictions are amfepramone and mazindol-containing drugs, GHB and its isomer or drugs that contain its sodium salt, phentermine, phendimetrazine, and propofol-containing drugs. The MFDS had already restricted approvals for the substances on August 14, 2020. One point of attention in the new announcement is that even narcotic medications subject to restrictions that were developed into a ‘new formulation’ may be subject to deliberations by the ‘Narcotics Safety Control Deliberation Committee’ if the MFDS determines the drug to have a high risk of misuse or abuse. Therefore, if the new formulation does not pass the ‘Narcotics Safety Control Deliberation review, that new drug may not receive marketing authorization in Korea. The MFDS announced such restrictions on narcotic medications due to intermittent applications filed for marketing authorizations by companies after changing only the formulation of their drugs to receive approval in categories where new marketing authorizations are restricted. As the authorities restricted new approvals in the above areas to eradicate misuse and abuse of narcotic medications, the MFDS needs to ponder whether to approve the new formulations when a pharmaceutical company requests approvals for developing new formulations. In the case of propofol, a restricted narcotic, the company applied for marketing authorization after changing its injection formulation into a prefilled syringe type. The drug was Fresenius Kabi’s ‘Fresofol MCT Prefilled Syringe.’ As a result, the MFDS issued a public notice and implemented an administrative measure to clearly prohibit approval of new narcotic medications with new formulations, etc., if the drug has a high possibility of misuse or abuse through. Also, by making the drugs subject to the Narcotics Safety Control Deliberation Committee review, the approvals of narcotic medications with new formulations will now be reviewed more professionally and objectively. In other words, narcotic medications that wish to obtain new approvals for their new formulations would need to have little risk of misuse or abuse and pass advisory review from the Narcotics Safety Control Deliberation Committee. An MFDS official said, “Among the narcotic medications that were restricted permission to minimize misuse and abuse, there were cases where companies sought new approvals based on new formulations. We made the announcement to clarify the need to restrict such approvals if their risk of misuse or abuse is high." The official added, “The notice further clarifies our regulations on how narcotic medications, even with new formulations, will not be approved if at high risk of misuse or abuse, or fails to pass deliberations by the Narcotics Safety Control Deliberation Committee.”
- Policy
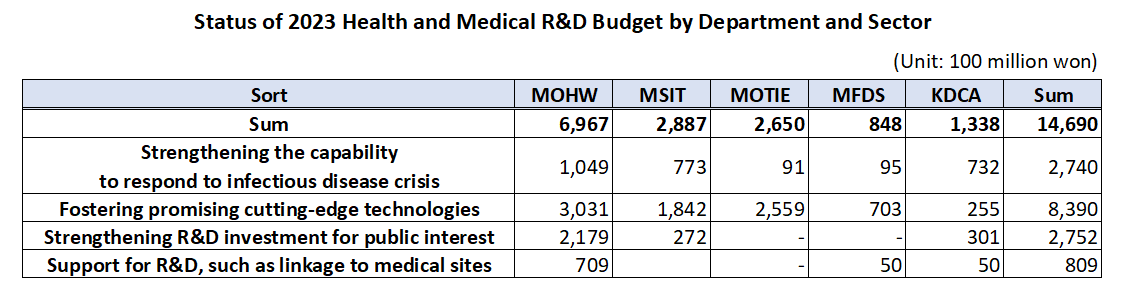
- Gov allocates KRW1.47 trillion budget for healthcare R&D
- by Kim, Jung-Ju Dec 30, 2022 06:33am
- The government’s budget for next year’s healthcare R&D including new drugs, medical devices, and digital transformation to AI-based biohealth is estimated to be around KRW 1.47 trillion. This is the total amount of budget that will be supported by the Ministry of Health and Welfare, Ministry of Science and ICT, Ministry of Trade, Industry, and Energy, Ministry of Food and Drug Safety, and Korea Disease Control and Prevention Agency, an amount similar to this year and 5% of the government’s total R&D budget. According to the MOHW on the 29th, the government plans to support KRW 149.5 billion in new tasks and KRW 1.32 trillion on ongoing tasks, for a total of 128 projects around 4 major areas including ▲reinforcing the ability to respond to infectious diseases, ▲fostering promising advanced technologies in biohealth, ▲increasing R&D investment for public interest including overcoming diseases, etc., and ▲support for R&D linked to the medical field. ◆Reinforcing ability to respond to infectious diseases = The government will be supporting KRW 274 billion for 26 select projects next year to strengthen Korea’s capacity in responding to infectious disease crises. The authorities plan to establish health security by supporting R&D to secure main technology capabilities for vaccines and treatments that can protect public lives and health from future infectious diseases. By ministry, the MOHW will be investing KRW 3.75 billion in developing antiviral treatments in preparation for RNA virus infections (Disease X), and MSIT has allocated KRW 13.3 billion for the Bio & Medical Technology Development Program and the establishment of the National Preclinical Trial Support System. The government also allocated a budget to advance the disease control and prevention system. Based on the lessons learned from the COVID-19 pandemic, the government plans to establish a safe society from new infectious diseases by promoting research on the whole disease control and prevention, etc to advance Korea’s disease control and prevention system. The MOHW allocated KRW 1.6 billion, KDCA KRW 1.3 billion, and MOTIE KRW 0.7 billion for the project to advance the pan-ministerial infectious disease control and prevention system. ◆Fostering promising advanced technologies= The government plans to support a total of KRW 839 billion in 66 projects to foster promising advanced technologies. First, the government will be improving the quality of healthcare by developing data and AI-based technology and prompting the digital transformation of biohealth, and providing personalized healthcare. In terms of new projects that will be initiated by each ministry, MOHW will be investing KRW 6.25 billion in R&D for technology to utilize healthcare MyData and its demonstration project. Also, the MOHW allocated KRW 7.5 billion to the project for developing healthcare technology based on virtual patients and hospitals, and KRW 7.5 billion for the demonstration and introduction of medical institution-based digital healthcare. The authorities will also be investing in the discovery of promising next-generation areas. It plans to continue the search for future drivers of biohealth by supporting R&D in unexplored areas such as microbiome, and in areas that can enhance industry competitiveness by improving the self-sufficiency of core technologies such as advanced medical devices. In terms of new projects that will be initiated by each ministry, the MOHW and KDCA allocated KRW 3.8 billion and KRW 1.3 billion each for the hospital-based human microbiome R&D project. The MFDS has also newly allocated KRW 7.4 billion to support regulatory science for pan-ministerial medical device regulations, and the MOTIE allocated KRW 2.1 billion for the development of interventional medical device technology based on advanced manufacturing technology. The projects also include R&D in regenerative medicine. The government plans to support research to establish a basis for the commercialization of regenerative medicine, such as xenotransplantation R&D projects, to ultimately become a global leader in advanced regenerative medicine technology by securing core and basic source technologies. In terms of new projects that will be initiated by each ministry, the MOHW will invest KRW 6 billion in xenotransplantation R&D projects, the MOTIE will invest KRW 4.5 billion in bio, medical technology development, and stem cell ATLA-based treatment technology for incurable diseases. ◆Strengthening R&D investment for public interest = The government will also allocate KRW 275.2 billion in 28 select projects to invest in R&D for public interest such as overcoming diseases. First, the government plans to ease the socioeconomic burden by focusing on the development of healthcare technologies to overcome diseases that bring high burden to the public such as brain diseases, mental health, and cancer. In terms of new projects that will be initiated by each ministry, the MOHW allocated KRW 4.95 billion in the development of technologies to address issues in the clinical field for diseases related to brain and nervous diseases, KRW 9.63 billion in an R&D project and its demonstration for the development of evidence-based personalized healthcare for cancer survivors, and KRW 0.5 billion for the development of technology for metaverse-based mental health management in the National Center for Mental Health. The government plans to preemptively conduct R&D in healthcare technologies where public demand is expected to surge in line with social changes including aging and the low birth rate, that will contribute to resolving social issues and enhancing the real sense of R&D in public health. For this, the MOHW will support KRW 3.9 billion in R&D of consumer-focused care robots and their service demonstration. ◆ support for R&D linked to the medical field= The government plans to support KRW 80.9 billion in R&D after selecting 8 projects linked with the medical field. First, it plans to reinforce Korea’s global competitiveness by fostering professional manpower that can drive innovation in biohealth, such as expanding research support for new physician-scientists and continuing training for regulatory science experts. For this, MOHW has allocated KRW 4.05 billion for the global research cooperation support project. Seong-ho Eun of the Bureau of Advanced Health Technology Policy MOHW said, “The government will continue to promote relevant policies and continue increasing R&D investments so that the healthcare R&D provided by the government serves as a basis for better quality healthcare services. We will also further activate the Health and Medical Technology Policy Deliberation Committee so that related ministries and the private sector for more organic cooperation and communication.”
- Policy
- A rapid change in the population
- by Kang, Shin-Kook Dec 29, 2022 06:03am
- Policy Tasks Determined at the Second Vice-Minister Meeting of the Ministry Related to Population Future Strategy. The government will come up with all-around measures due to population changes caused by the world's highest rate of low birth rate and aging society. This included institutionalization of non-face-to-face treatment and visiting medical services. On the 28th, the government held the second vice-ministerial meeting of ministries related to population future strategy presided over by Na Kyung-won, vice chairman of the Low Birth Rate and Aging Society Committee, and announced "Demographic Change and Countermeasures." This is because the population decreased by 37.66 million in 2070 due to low birth rates, and side effects such as a surge in the elderly population and the disappearance of the region were imminent. ◆ Non-face-to-face treatment and medical-care supply = In order to improve medical access to books and wallpaper and improve patient health, it will also promote the institutionalization of non-face-to-face treatment centered on primary medical institutions next year. In addition, a clinic-level medical institution that provides visiting medical care and care services will be designated and a pilot project for a home medical center will be implemented this month. In addition, the "contract doctor system" that regularly visits nursing facilities where doctors do not reside to check and manage the health status of patients will be enhanced. ▲ A schedule for promoting the health and welfare sector among major tasks in response to the population crisis The government will also review measures to expand supply by supporting the private sector's entry into the elderly care service sector and inducing diversification and scale. It aims to create a foundation for expanding the private supply of various services by introducing a self-burden system for elderly customized care services and providing universal services. ◆ Regional medical personnel and health insurance financial efficiency = Consultations on adjustment of the number of medical schools to cope with the increase in medical demand due to the expansion of training at local hospitals and aging of majors will also begin. Currently, the medical school has a quota of 3,058. In addition, a pilot project for joint training between national university hospitals and local medical centers will begin in March next year. The government plans to establish a "fiscal vision 2050" in the first half of next year to discover reform tasks for overall economic and social structural problems such as responding to population decline. It supports the discussion of pension reform measures by the National Pension Reform Special Committee and also prepares measures to preemptively streamline health insurance spending. Various pilot projects will be promoted to convert the value-based payment system based on medical service performance, not input, and appropriate medical use inducement measures will be prepared, such as rationalizing the use of non-benefits and linking public health insurance. Disclosure of non-benefits, non-benefit reporting by medical institutions, and establishment of a health insurance-loss insurance-related management system are expected to be on the agenda. "The world's fastest-growing demographic change will have a wide impact on the economy and society, including education, military service, local economy, growth potential, industrial structure, and welfare system," a government official said. "In the short term, the growth potential is weakened due to the aging population."
- Policy
- Takeda CMV infection treatment Livtencity has been approved
- by Kim, Jung-Ju Dec 29, 2022 06:03am
- After transplantation of Takeda Pharmaceutical Korea, Cytomegalovirus infection treatment Livtencity obtained domestic item permission and passed the first gateway to supply. The Ministry of Food and Drug Safety (Director Oh Yoo-kyung) announced on the 27th that it has approved Livtencity of Takeda, a rare drug. Cytomegalovirus (CMV) is asymptomatic and is reactivated, causing serious diseases. Livticity is an oral antiviral drug that inhibits the growth of the virus by lowering the activity of the "UL97 protein phosphorylation enzyme" involved in cloning and proliferation in CMV. The drug is expected to provide new treatment opportunities for adult patients with macrophage virus infection after transplantation, which is resistant to or unresponsive to one or more of the existing antiviral drugs Ganciclovir, Valganciclovir, Foscavir, and Cidofovir.